845614-11-1
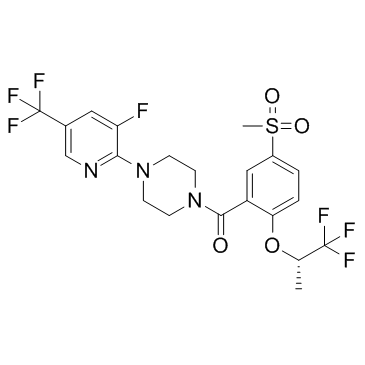
845614-11-1 structure
- Name: Bitopertin
- Chemical Name: [4-[3-fluoro-5-(trifluoromethyl)pyridin-2-yl]piperazin-1-yl]-[5-methylsulfonyl-2-[(2S)-1,1,1-trifluoropropan-2-yl]oxyphenyl]methanone
- CAS Number: 845614-11-1
- Molecular Formula: C21H20F7N3O4S
- Molecular Weight: 543.45500
- Catalog: Signaling Pathways Membrane Transporter/Ion Channel GlyT
- Create Date: 2019-01-03 20:08:18
- Modify Date: 2024-01-02 12:39:37
-
Bitopertin is a potent, noncompetitive glycine reuptake inhibitor, inhibits glycine uptake at human GlyT1 with a concentration exhibiting IC50 of 25 nM.
| Name | [4-[3-fluoro-5-(trifluoromethyl)pyridin-2-yl]piperazin-1-yl]-[5-methylsulfonyl-2-[(2S)-1,1,1-trifluoropropan-2-yl]oxyphenyl]methanone |
|---|---|
| Synonyms |
[4-(3-fluoro-5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-[5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methyl-ethoxy)-phenyl]-methanone
RG 1678 Paliflutine Paliflutine [INN] (S)-[4-(3-fluoro-5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-[5-methanesulfonyl-2-(2,2,2-trifluoro-1-methyl-ethoxy)-phenyl]-methanone Bitopertin |
| Description | Bitopertin is a potent, noncompetitive glycine reuptake inhibitor, inhibits glycine uptake at human GlyT1 with a concentration exhibiting IC50 of 25 nM. |
|---|---|
| Related Catalog | |
| Target |
IC50: 25 nM (GlyT1)[1] |
| In Vitro | Bitopertin (RG1678) competitively blocks [3H]ORG24598 binding sites at human GlyT1b in membranes from Chinese hamster ovary cells. Bitopertin potently inhibits [3H]glycine uptake in cells stably expressing hGlyT1b and mGlyT1b, with IC50 values of 25±2 nM and 22±5 nM, respectively (n=6). Conversely, Bitopertin has no effect on hGlyT2-mediated glycine uptake up to 30 μM concentration. Bitopertin has high affinity for the recombinant hGlyT1b transporter. Under equilibrium conditions (1 h at room temperature), Bitopertin displaces [3H]ORG24598 binding with a Ki of 8.1 nM. In hippocampal CA1 pyramidal cells, Bitopertin enhances NMDA-dependent long-term potentiation at 100 nM but not at 300 nM[1]. Additional profiling revealed that Bitopertin (RG1678) has an excellent selectivity profile against the GlyT2 isoform (IC50>30 μM) and toward a panel of 86 targets including transmembrane and soluble receptors, enzymes, ion channels, and monoamine transporters (<41% inhibition at 10 μM is measured for all targets)[2]. |
| In Vivo | Bitopertin (RG1678) dose-dependently increases cerebrospinal fluid and striatal levels of glycine measured bymicrodialysis in rats. Additionally Bitopertin attenuates hyperlocomotion induced by the psychostimulant D-amphetamine or the NMDA receptor glycine site antagonist L-687,414 in mice. Bitopertin also prevents the hyper-response to D-amphetamine challenge in rats treated chronically with phencyclidine, an NMDA receptor open-channel blocker. Administration of vehicle has no effect on extracellular levels of striatal glycine, which remained constant throughout the experiment. In contrast, p.o. administration of Bitopertin (1-30 mg/kg) produced a dose-dependent increase in extracellular glycine levels. Bitopertin 30 mg/kg produces glycine levels 2.5 times higher than pretreatment levels. A similar dose-dependent increase in glycine concentration is observed in the CSF of rats treated p.o. with Bitopertin (1-10 mg/kg) compared with vehicle-treated animals, 3 h after drug administration. Interestingly, the level of CSF glycine increase 3 h after Bitopertin dosing is very similar to the increase in the microdialysis experiment at the same time point[1]. In vivo pharmacokinetic studies in rat and monkey reveals that Bitopertin (RG1678) has, in both species, a low plasma clearance, an intermediate volume of distribution, a good oral bioavailability (78% for rat, 56% for monkey), and a favorable terminal half-life (5.8 h for rat, 6.4 h for monkey). The plasma protein binding is high in the two preclinical species (97%) and in human (98%). The CNS penetration of Bitopertin in rat (brain/plasma=0.7) is better than that in mouse (brain/plasma=0.5)[2]. |
| Kinase Assay | Association and dissociation kinetic analysis of [3H]ORG24598 to hGlyT1 and ratforebrain membranes is performed. [3H]ORG24598 binding experiments are performed using membranes from CHO cells expressing hGlyT1b and also in membranes from mouse, rat, monkey, and dogforebrains. Saturation isotherms are determined by adding [3H]ORG24598 to rat, mouse, monkey, and dog forebrain membranes (40 μg/well) and cell membranes (10 μg/well) in a total volume of 500 μL for 3 h at room temperature. Saturation binding experiments are analyzed by an Excel-based curve-fitting program using the Michaelis-Menten equation derived from the equation of a bimolecular reaction and the law of mass action:B=(Bmax×[F])/(Kd+[F]), where B is the amount of ligand bound at equilibrium, Bmax the maximum number of binding sites, [F] the concentration of free ligand, and Kd the ligand dissociation constant. For inhibition experiments, membranes are incubated with 3 nM [3H]ORG24598 and 10 concentrations of Bitopertin for 1 h at room temperature. Schild analysis is performed in the presence of increasing concentrations of [3H]ORG24598 (1-300 nM). IC50 values are derived as described above. Ki values are calculated according to the following equation: Ki=IC50/(1+[L]/Kd)[1]. |
| Animal Admin | Mice[1] Male NMRI mice (20-30 g) are treated with Bitopertin (0.3, 3, 1, and 10 mg/kg p.o.) or vehicle (p.o.). After 1 min, L-687,414 (50 mg/kg s.c.) or vehicle is given. After 15 min of habituation in the activity chambers, horizontal activity is recorded for 60 min. The time course of Bitopertin effects on L-678,414-induced hyperactivity is also examined; locomotor activity is assessed 2.5, 4.5, and 24 h after administration of Bitopertin (L-678,414 is always given 15 min before the activity procedure). In addition, the effect of subchronic Bitopertin is investigated. Mice receive vehicle or Bitopertin (1 mg/kg p.o.) for 4 consecutive days and L-678,414-induced hyperactivity is evaluated on day 5. Rats[1] Wistar rats receive a 14-day treatment of PCP HC1 (5 mg/kg) or vehicle (NaCl 0.9%, 5 mL/kg i.p.). 24 h following the last injection, rats (6-18 per group) are allowed to individually habituate to the test boxes for 30 min. Rats then received Bitopertin (1, 3, 10 mg/kg p.o.) or vehicle (Polysorbate 80, HEC, Methyl- and Propylparaben pH 6.0; 5 mL/kg p.o.), followed after 1 h by 1 mg/kg D-amphetamine or vehicle i.p. Horizontal activity is recorded directly after the administration of Bitopertin until 120 min after dosing with amphetamine. Data are analyzed by ANOVA supplemented by Fischer's least significant difference post hoc test. |
| References |
| Molecular Formula | C21H20F7N3O4S |
|---|---|
| Molecular Weight | 543.45500 |
| Exact Mass | 543.10600 |
| PSA | 88.19000 |
| LogP | 5.01870 |
| Storage condition | -20°C |
|
~79%
845614-11-1 |
| Literature: Pinard, Emmanuel; Alanine, Alexander; Alberati, Daniela; Bender, Markus; Borroni, Edilio; Bourdeaux, Patrick; Brom, Virginie; Burner, Serge; Fischer, Holger; Hainzl, Dominik; Halm, Remy; Hauser, Nicole; Jolidon, Synese; Lengyel, Judith; Marty, Hans-Peter; Meyer, Thierry; Moreau, Jean-Luc; Mory, Roland; Narquizian, Robert; Nettekoven, Mathias; Norcross, Roger D.; Puellmann, Bernd; Schmid, Philipp; Schmitt, Sebastien; Stalder, Henri; Wermuth, Roger; Wettstein, Joseph G.; Zimmerli, Daniel Journal of Medicinal Chemistry, 2010 , vol. 53, # 12 p. 4603 - 4614 |
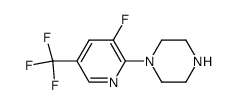
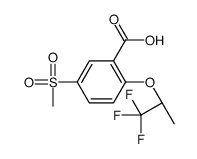
| Precursor 2 | |
|---|---|
| DownStream 0 | |