PLX8394
更新时间:2024-01-08 18:27:16
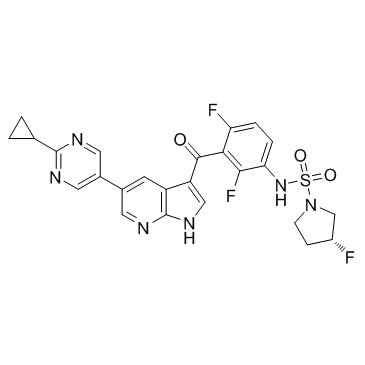
PLX8394结构式

|
常用名 | PLX8394 | 英文名 | PLX8394 |
|---|---|---|---|---|
| CAS号 | 1393466-87-9 | 分子量 | 542.533 | |
| 密度 | 1.6±0.1 g/cm3 | 沸点 | 786.8±70.0 °C at 760 mmHg | |
| 分子式 | C25H21F3N6O3S | 熔点 | N/A | |
| MSDS | N/A | 闪点 | 429.6±35.7 °C |
PLX8394用途PLX8394 是一种有效的,选择性的 Raf 抑制剂,能够抑制 BRAFV600E 的活性,IC50 值约为 5 nM。 |

| 中文名 | PLX8394 |
|---|---|
| 英文名 | plx8394 |
| 英文别名 | 更多 |
| 描述 | PLX8394 是一种有效的,选择性的 Raf 抑制剂,抑制 BRAFV600E 活性的 IC50 值约为 5 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
BRafV600E:5 nM (IC50) |
| 体外研究 | PLX8394在> 25nM下有效抑制PRT#3和PRT#4细胞中ERK1/2的磷酸化,并且除了10nM的亲本细胞外还有效抑制ERK1/2的磷酸化。 PLX8394有效减少细胞周期蛋白D3和细胞周期蛋白D1,视网膜母细胞瘤蛋白的磷酸化,以及亲代细胞和PRT#3和PRT#4细胞中细胞周期蛋白A2的表达。 PLX8394抑制ERK1/2磷酸化和vemurafenib抗性细胞的生长,所述细胞携带BRAF V600K/L505H双突变或转座子诱导的N末端截短形式的BRAF [1]。 PLX8394显着损害肿瘤细胞生长并抑制表达内源性V600E或非V600突变形式BRAF的LA细胞系中的MAPK信号传导[2]。 |
| 体内研究 | PLX8394(150mg/kg/d)基本上抑制这些H1755异种移植肿瘤中的肿瘤生长,MAPK途径信号传导和肿瘤细胞增殖,而在小鼠中没有明显的毒性。 PLX8394与厄洛替尼联合使血浆厄洛替尼浓度>1μM[2]。 |
| 细胞实验 | 对于MTT测定,将2×103个细胞在其常规培养基(含有PRX系的PLX4720)的96孔中一式三份接种。第二天,用PBS洗涤细胞两次,然后补充含有指定的RAF抑制剂的培养基。 48小时后更换培养基,再过48小时后,向孔中加入10μL5mg/ mL MTT试剂,并培养3小时。然后将Formazan晶体用1:10稀释的0.1M甘氨酸(pH 10.5)在DMSO中溶解过夜。然后在Multiskan®频谱分光光度计中以450nM分析孔。所描绘的结果归一化为DMSO条件,并且是三个独立实验的复合物。显示的误差棒代表平均值的标准误差(SEM)。 |
| 动物实验 | 通过将50×50混合物中的5×10 6个细胞注射到基质胶和PBS中,产生H1755肿瘤异种移植物,产生6至8周龄的雌性NOD / SCID小鼠。一旦肿瘤达到150mm 3的平均大小,将小鼠随机分配到治疗组。 sc植入H1755细胞并使其生长至约200mm 3(植入后4周)。然后用载体,vemurafenib或PLX8394处理小鼠15天。每日口服强饲法的载体是PEG 400 [20%(体积/体积)],生育酚聚乙二醇琥珀酸酯(TPGS)[5%(体积/体积)],水[75%(体积/体积)]。将PLX8394溶解于PEG 400 [20%(体积/体积)],TPGS [5%(体积/体积)]和水[75%(体积/体积)]中并在整个给药期间连续涡旋。每天通过口服强饲法给予PLX8394,剂量为150mg / kg /天。 |
| 参考文献 |
| 密度 | 1.6±0.1 g/cm3 |
|---|---|
| 沸点 | 786.8±70.0 °C at 760 mmHg |
| 分子式 | C25H21F3N6O3S |
| 分子量 | 542.533 |
| 闪点 | 429.6±35.7 °C |
| 精确质量 | 542.134766 |
| LogP | 0.97 |
| 外观性状 | 粉末 |
| 蒸汽压 | 0.0±2.7 mmHg at 25°C |
| 折射率 | 1.703 |
| 储存条件 | -20℃ |
| 1-Pyrrolidinesulfonamide, N-[3-[[5-(2-cyclopropyl-5-pyrimidinyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl]-2,4-difluorophenyl]-3-fluoro-, (3R)- |
| (3R)-N-(3-{[5-(2-Cyclopropyl-5-pyrimidinyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl}-2,4-difluorophenyl)-3-fluoro-1-pyrrolidinesulfonamide |

- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:PLX8394游离态
- 纯度:98.22%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:PLX8394
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]PLX8394,97%
- 产品网址:https://www.canspec.cn/product/816237.html
- 纯度:97.0%
- 规格:5mg
- 联系人:夏言
- 联系电话:18016376636
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:PLX8394
- 产品网址:https://www.aladdin-e.com/zh_cn/P413892.html
- 纯度:97.0%
- 规格:1mg/25mg/5mg/1g
- 联系人:阿拉丁
- 联系电话:400-620-6333
- 武汉壹加壹生物科技有限公司
- 湖北省 武汉市 江夏区
- 产品名:PLX8394
- 纯度:98.0%
- 规格:1g
- 联系人:刘经理
- 联系电话:16602739303
查看所有供应商和价格请点击: