PZ-1
更新时间:2024-01-16 18:20:10
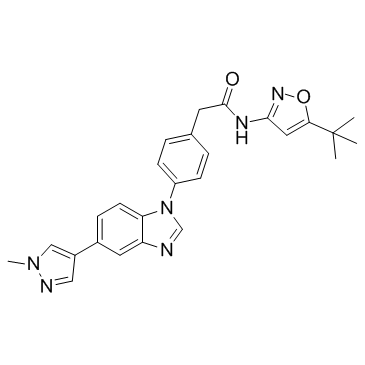
PZ-1结构式

|
常用名 | PZ-1 | 英文名 | Pz-1 |
|---|---|---|---|---|
| CAS号 | 1800505-64-9 | 分子量 | 454.52 | |
| 密度 | N/A | 沸点 | N/A | |
| 分子式 | C26H26N6O2 | 熔点 | N/A | |
| MSDS | N/A | 闪点 | N/A |
PZ-1用途Pz-1是有效地 RET 和 VEGFR2 受体酪氨酸激酶抑制剂,Pz-1抑制这两个野生型激酶的 IC50 值小于 1 nM。 |

| 中文名 | PZ-1 |
|---|---|
| 英文名 | Pz-1 |
| 描述 | Pz-1是有效地 RET 和 VEGFR2 受体酪氨酸激酶抑制剂,Pz-1抑制这两个野生型激酶的 IC50 值小于 1 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: < 1 nM (RET and VEGFR2)[1] |
| 体外研究 | Pz-1是II型酪氨酸激酶抑制剂,能够结合激酶的DFG外构象。在基于细胞的分析中,1.0 nM的Pz-1强烈抑制VEGFR2的酪氨酸磷酸化和临床相关的RET突变体,包括vandetanib和cabozantinib难治的那些(RETV804M和RETV804L)[1]。 |
| 体内研究 | Pz-1在VEGFR2上显示有活性,可阻断RET刺激生长所需的血液供应。在口服1.0mg/kg /天时,Pz-1消除由RET-突变体成纤维细胞诱导的肿瘤形成,并阻断肿瘤组织中RET和VEGFR2的磷酸化。 Pz-1在100.0 mg/kg时没有可检测到的毒性,这表明治疗窗口很大[1]。 |
| 动物实验 | 小鼠[1]为了解决体内RET驱动的作用,与组成型活性对照Ras癌基因相比,对由致癌RET诱导的肿瘤测试Pz-1。然后向免疫缺陷(nu / nu)小鼠注射NIH3T3 RETC634Y或NIH3T3 HRasG12V细胞,并且在出现肿瘤之前,用Pz-1(1.0,3.0或10.0mg / kg /天)处理PO或不进行处理。然后测量肿瘤效应[1] |
| 参考文献 |
| 分子式 | C26H26N6O2 |
|---|---|
| 分子量 | 454.52 |
| 储存条件 | -20°C,密闭,干燥 |

- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:Pz-1
- 纯度:99.95%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:Pz-1
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]Pz-1,98%
- 产品网址:https://www.canspec.cn/product/831115.html
- 纯度:98.0%
- 规格:5mg
- 联系人:夏言
- 联系电话:18016376636
查看所有供应商和价格请点击: