718630-59-2
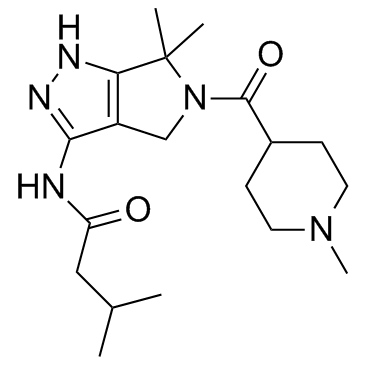
718630-59-2 structure
- Name: PHA-793887
- Chemical Name: N-[6,6-dimethyl-5-(1-methylpiperidine-4-carbonyl)-1,4-dihydropyrrolo[3,4-c]pyrazol-3-yl]-3-methylbutanamide
- CAS Number: 718630-59-2
- Molecular Formula: C19H31N5O2
- Molecular Weight: 361.482
- Catalog: Biochemical Inhibitor Cell Cycle CDK inhibitor
- Create Date: 2018-12-22 19:04:10
- Modify Date: 2026-07-07 18:04:01
-
PHA-793887 is a potent, ATP-competitive CDK inhibitor, can inhibit Cdk2, Cdk1, Cdk4, and Cdk9 with IC50s of 8 nM, 60 nM, 62 nM and 138 nM, respectively, and also inhibits glycogen synthase kinase 3β with an IC50 of 79 nM.
| Name | N-[6,6-dimethyl-5-(1-methylpiperidine-4-carbonyl)-1,4-dihydropyrrolo[3,4-c]pyrazol-3-yl]-3-methylbutanamide |
|---|---|
| Synonyms |
N-{6,6-Dimethyl-5-[(1-methyl-4-piperidinyl)carbonyl]-1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazol-3-yl}-3-methylbutanamide
N-(6,6-Dimethyl-5-(1-methylpiperidine-4-carbonyl)-1,4,5,6-tetrahydropyrrolo(3,4-C)pyrazol-3-yl)-3-methylbutanamide S1487_Selleck 3-Methyl-N-[1,4,5,6-tetrahydro-6,6-dimethyl-5-[(1-methyl-4-piperidinyl)carbonyl]pyrrolo[3,4-c]pyrazol-3-yl]butanamide PHA-793887 |
| Description | PHA-793887 is a potent, ATP-competitive CDK inhibitor, can inhibit Cdk2, Cdk1, Cdk4, and Cdk9 with IC50s of 8 nM, 60 nM, 62 nM and 138 nM, respectively, and also inhibits glycogen synthase kinase 3β with an IC50 of 79 nM. |
|---|---|
| Related Catalog | |
| Target |
Cdk5/p25:5 nM (IC50) cdk2/cyclin A:8 nM (IC50) CDK2/cyclinE:8 nM (IC50) CDK7/cyclin H:10 nM (IC50) Cdk1/cyclin B:60 nM (IC50) Cdk4/cyclin D1:62 nM (IC50) CDK9/cyclinT1:138 nM (IC50) GSK-3β:79 nM (IC50) |
| In Vitro | PHA-793887 partially inhibits Rb phosphorylation at 1 μM and almost completely at 3 μM, in A2780 tumor cell line. PHA-793887 (1 μM) partially inhibits phosphorylation of the Cdk2 substrates Rb and NPM in A2780 tumor cell line. PHA-793887 (6 μM) significantly inhibits Rb and NPM phosphorylation in MCF7 cell line[1]. PHA-793887 shows cytotoxic activities against leukemic cell lines in vitro, with IC50 ranging from 0.3 to 7 μM. In colony assays, PHA-793887 is highly cytotoxic for leukemia cell lines, with an IC50 <0.1 μM. PHA-793887 induces cell-cycle arrest, inhibits Rb and nucleophosmin phosphorylation, and modulates cyclin E and cdc6 expression at low doses of 0.2 to 1 μM and induces apoptosis at the highest dose of 5 μM. PHA-793887 is a novel inhibitor of several cdk, including cdk1, cdk2, cdk4, cdk5, cdk7, and cdk9 with IC50 in the 5 to 140 nM range[3]. |
| In Vivo | PHA-793887 induces tumor growth inhibition in the range of 50% at dose of 15 mg/kg to 75% at dose of 30 mg/kg in CD-1 nude mice. PHA-793887 (30 mg/kg, i.v.) also induces significant downregulation of the 58-gene panel in the skin of CD-1 mice[1]. PHA-793887 (20 mg/kg, i.v.) induces tumor regression in the HL60 model. In the K562 model, PHA-793887 significantly reduces tumor growth. Moreover, PHA-793887 (20 mg/kg, i.v.) inhibits human primary leukemia growth in engraftment setting in vivo[3]. |
| Cell Assay | Cytotoxicity assays are performed using the Alamar blue vital dye. For each cell line, preliminary dose−response curves are performed to establish the cell-concentration range, giving a linear relationship with fluorescence. For cell lines, 5,000 to 20,000 cells are plated in 200 μL complete medium in 96-well plates, in the presence or absence of increasing doses of drugs (0.01−10 μM). For ALL-2 and AML-PS leukemias 10 × 105 cells/well are plated in StemSpanSFEM medium and treated with the same range of drug concentrations. Peripheral blood mononuclear cells and cord blood CD34+ cells are plated 1 × 105 cells/well in presence or absence of 1 μg/mL phytohemagglutin or growth factor cocktail (50 ng/mL stem cell factor, 20 ng/mL each of granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, interleukin-3, interleukin-6, and 3 U/mL erythropoietin), respectively. In all cases, after 48 hours culture, 1/10 volume Alamar blue solution is added and incubated overnight. The plates are then read in a fluorimeter with excitation at 535 nm and emission at 590 nm. Cytotoxicity is calculated as percentage of fluorescence with respect to untreated control, after subtracting for background fluorescence in absence of cells. |
| Animal Admin | 107 HL60 and K562 cells are inoculated subcutaneously in SCID mice. Animals are randomized in seven mice per group. PHA-793887 is administered at 20 mg/kg intravenous (IV) once a day, continuously for 10 days (from day 9 to day 18) in HL60 model and with a two 5-day cycles (from day 9 to day 13 and from day 17 to day 21) in K562-bearing mice. Glivec is orally administered for 9 consecutive days from day 9 onward in the K562 xenograft model. Tumor growth and net body weight are evaluated twice a week. The tumor weight is calculated according to the following formula: tumor weight = length (mm) × width2 (mm) /2. The effect of the anticancer treatment is determined as the delay in onset of an exponential growth of tumors. This delay (T − C value) is defined as the difference of median time (in days) required for the tumors of treatment (T) and control groups (C) to reach a predetermined size. Toxicity is evaluated on the basis of the body weight reduction. |
| References |
| Density | 1.2±0.1 g/cm3 |
|---|---|
| Boiling Point | 596.2±50.0 °C at 760 mmHg |
| Molecular Formula | C19H31N5O2 |
| Molecular Weight | 361.482 |
| Flash Point | 314.4±30.1 °C |
| Exact Mass | 361.247772 |
| PSA | 84.82000 |
| LogP | 1.79 |
| Vapour Pressure | 0.0±1.7 mmHg at 25°C |
| Index of Refraction | 1.573 |
| Storage condition | -20°C |