| Description |
Lonafarnib is an orally bioavailable farnesyl protein transferase (FPTase) inhibitor for H-ras, K-ras and N-ras with IC50 of 1.9 nM, 5.2 nM and 2.8 nM, respectively.
|
| Related Catalog |
|
| Target |
IC50: 1.9 nM (H-ras), 5.2 nM (K-ras), 2.8 nM (N-ras)[1]
|
| In Vitro |
Lonafarnib (Sch66336) potently inhibits Ha-Ras processing in whole cells and blocks the trans formed growth properties of fibroblasts and human tumor cell lines expressing activated Ki-Ras proteins[1]. All treatment groups containing Lonafarnib (10 µM) show a significantly higher level of unfarnesylated H-Ras (116-137%) compared to control treatment[2].
|
| In Vivo |
In mouse, rat, and monkey systems, Lonafarnib (Sch66336) has excellent oral bioavailability and pharmacokinetic properties. In the nude mouse, Lonafarnib demonstrates potent oral activity in a wide array of human tumor xenograft models including tumors of colon, lung, pancreas, prostate, and urinary bladder origin[1]. Lonafarnib alone (80 mg/kg by oral gavage, once daily) has limited ability to inhibit orthotopic U87 tumors compared to vehicle treated control animals (T/C of 0.67). The combination of XRT/Tem (2.5Gy/day for 2 days; 5 mg/kg by oral gavage 90 min prior to XRT) is designed to produce modest tumor growth inhibition in vivo(T/C of 0.42). Concurrent Lonafarnib/XRT/Tem (Lonafarnib 80 mg/kg by oral gavage, once daily, XRT 2.5Gy/day for 2 days, and Tem 5 mg/kg by oral gavage 90 min prior to XRT) provides the strongest growth reduction (T/C of 0.02) and is significantly more effective than XRT/Tem (p<0.04), with the majority of animals demonstrating a decrease in tumor volume (p<0.05) after two weeks and persisting after 4 weeks (p<0.05)[2].
|
| Kinase Assay |
FPTactivity is determined by measuring the transfer of [3H]farnesyl from [3H]farnesyl PPi to trichloroacetic acid-precipitable Ha-Ras-CVLS. GGPT-1 activity is similarly determined using [3H]geranylgeranyl diphosphate and Ha-Ras-CVLL as substrates[1].
|
| Cell Assay |
CellTiter96 Aqueous Assay kit is used. Assays are performed with 5000 cells/well in a 96-well tissue culture plate. Plates are irradiated 24 h after drug exposure and assayed 96 h after XRT, with fresh drug treatments applied each day. For quantification, dye is added directly to each well, plates are washed as per the manufactures recommendation and cell viability determined by optical density. Significance is analyzed using the Student’s T-test. 12-well plates are seeded with 100,000 cells/well. Drug treatments are initiated 24 h after plating, and media is replaced every 24 h for a total of 96 h of drug exposure. Plates are irradiated after 24 h of drug exposure. Cells from triplicate sets of treatments are trypsonized and counted 48 h after irradation using a Z1 series coulter counter, and compared to cell numbers from wells counted on Day 1 (the day drug treatment is initiated). Proliferation after drug treatments are normalized to the control wells and expressed as % of the control treatment. Significance is analyzed using the Student’s T-test[2].
|
| Animal Admin |
Mice[2] Lonafarnib is given once daily at 80mg/kg with twice weekly weightings to ensure accurate dosing. Temozolomide (Tem) is given by gavage at 5 mg/kg 90 min prior to XRT. For irradiation, anesthetized mice are placed in a lead shielding apparatus which limited radiation exposure to the head only. Treatment (2.5Gy/day for two days) is delivered using a Gammacell 40 irradiator delivering 100 rads/min. For in vivo combination experiments, suboptimal doses of XRT/Tem are selected to permit identification of synergistic effects of Lonafarnib.
|
| References |
[1]. Liu M, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998 Nov 1;58(21):4947-56. [2]. Chaponis D, et al. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J Neurooncol. 2011 Aug;104(1):179-89.
|
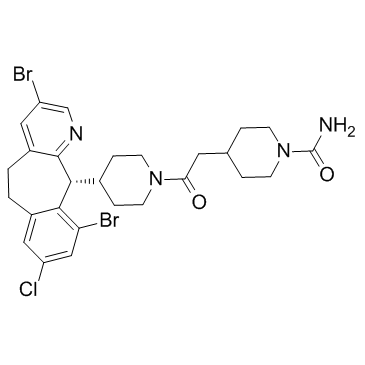

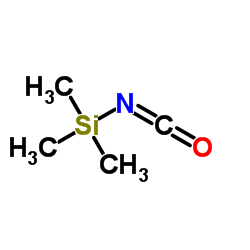 CAS#:1118-02-1
CAS#:1118-02-1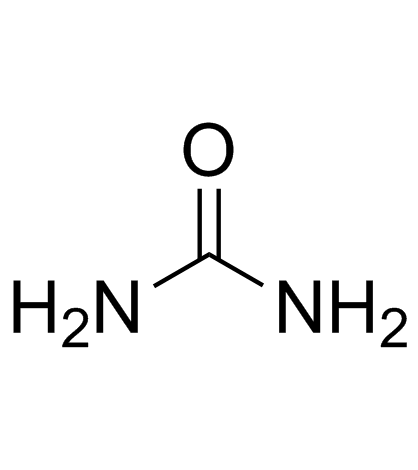 CAS#:57-13-6
CAS#:57-13-6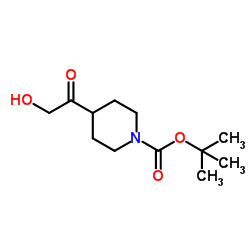 CAS#:157688-46-5
CAS#:157688-46-5![5H-Benzo[5,6]cyclohepta[1,2-b]pyridine, 3,10-dibromo-8-chloro-6,11-dihydro-11-(4-piperidinyl)-, (11R)- Structure](https://image.chemsrc.com/caspic/017/193276-49-2.png) CAS#:193276-49-2
CAS#:193276-49-2![(+)-1,1-dimethylethyl[[[4-(8-chloro-3,10-dibromo-6,11-dihydro-5H-benzo-[5,6]cyclohepta[1,2-b]pyridin-11(R)-yl)-1-piperidinyl]-carbonyl]-methyl]-1-piperidinecarboxylate Structure](https://image.chemsrc.com/caspic/010/193276-76-5.png) CAS#:193276-76-5
CAS#:193276-76-5![4-[3,10-dibromo-8-chloro-5,6-dihydro-11H-benzo-[5,6]cyclohepta[1,2-b]pyridin-11-ylidene]-1-piperidine Structure](https://image.chemsrc.com/caspic/093/193276-47-0.png) CAS#:193276-47-0
CAS#:193276-47-0![4-[3,10-dibromo-8-chloro-5,6-dihydro-11H-benzo-[5,6]cyclohepta[1,2-b]pyridin-11-ylidene]-1-piperidinecarboxylic acid ethyl ester Structure](https://image.chemsrc.com/caspic/141/193276-46-9.png) CAS#:193276-46-9
CAS#:193276-46-9