
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:N-(2-(diMethylaMino)ethyl)-1-(3-((4-((2-Methyl-1H-indol-5-yl)oxy)pyriMidin-2-yl)aMino)phenyl)MethanesulfonaMide
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:Sulfatinib
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:Sulfatinib
- 纯度:98.0%
- 规格:1mg
- 联系人:李先生
- 联系电话:18916172912
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:N-(2-(二甲基甲基)乙基)-1-(3-((4-((2-甲基-1H-吲哚-5-基)氧基)吡啶咪丁-2-基)氨基)苯基)甲烷磺酰亚胺
- 纯度:98.0%
- 规格:5mg/100mg/10mg/25mg
- 联系人:阿拉丁
- 联系电话:400-620-6333
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]N-(2-(diMethylaMino)ethyl)-1-(3-((4-((2-Methyl-1H-indol-5-yl)oxy)pyriMidin-2-yl)aMino)phenyl)MethanesulfonaMide,98%
- 纯度:98.0%
- 规格:200mg/1g/5g
- 联系人:夏言
- 联系电话:18016376636
查看所有供应商和价格请点击:
1308672-74-3
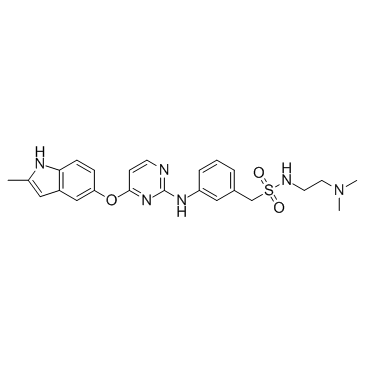
1308672-74-3结构式
- 常用中文名:Sulfatinib
- 常用英文名:Sulfatinib
- CAS号:1308672-74-3
- 分子式:C24H28N6O3S
- 分子量:480.582
- 相关类别: 研究领域 癌症
- 发布时间:2018-01-06 13:33:05
- 更新时间:2024-01-05 06:34:44
-
Sulfatinib (HMPL-012) 是一种有效且高度选择性的针对 VEGFR1/2/3,FGFR1和 CSF1R 的酪氨酸激酶抑制剂,IC50 值在1 到 24 nM之间。
| 中文名 | Sulfatinib |
|---|---|
| 英文名 | Sulfatinib |
| 英文别名 |
N-[2-(Dimethylamino)ethyl]-1-[3-({4-[(2-methyl-1H-indol-5-yl)oxy]-2-pyrimidinyl}amino)phenyl]methanesulfonamide
Benzenemethanesulfonamide, N-[2-(dimethylamino)ethyl]-3-[[4-[(2-methyl-1H-indol-5-yl)oxy]-2-pyrimidinyl]amino]- |
| 描述 | Sulfatinib (HMPL-012) 是一种有效且高度选择性的针对 VEGFR1/2/3,FGFR1和 CSF1R 的酪氨酸激酶抑制剂,IC50 值在1 到 24 nM之间。 |
|---|---|
| 相关类别 | |
| 靶点 |
VEGFR1 VEGFR2 VEGFR3 FGFR1 CSF1R |
| 体外研究 | Sulfatinib抑制VEGFR1,2和3,FGFR1和CSF1R激酶,IC50为1~24 nM,强烈阻断HEK293KDR细胞中VEGF诱导的VEGFR2磷酸化,CSF1刺激RAW264.7细胞中CSF1R磷酸化,IC50为2,分别为79 nM。 Sulfatinib还可以减弱VEGF或FGF刺激的HUVEC细胞增殖,IC50 <50 nM [1]。此外,它是一种hERG抑制剂,在CHO细胞中IC50为6.8μM[2]。 |
| 体内研究 | 在动物研究中,单次口服给予硫达肝素以暴露依赖性方式抑制VEGF刺激裸鼠肺组织中的VEGFR2磷酸化。此外,给药后24小时血浆中FGF23水平的升高提示抑制FGFR信号传导。 Sulfatinib在多种人异种移植模型中显示出有效的肿瘤生长抑制并显着降低CD31表达,表明通过VEGFR和FGFR信号传导对血管生成具有强烈抑制作用。在同源小鼠结肠癌模型CT-26中,Sulfatinib在单药治疗后表现出中度肿瘤生长抑制[1]。口服10 mg/kg后,小鼠的AUC和Cmax分别为397 ng/mL和138ng/mL [1]。 |
| 激酶实验 | 使用Z-lyte测定试剂盒测试KDR激酶抑制活性。测试系统在384孔板中含有300ng / mL重组人KDR催化结构域,10μMATP,1μM底物肽和一系列不同浓度的测试化合物(Sulfatinib);总体积为10μL。酶抑制在室温(25℃)下在室温下在振荡器上进行1小时。加入5μL终止溶液以终止反应[2]。 |
| 动物实验 | 在单次静脉内和口服2.5和10mg / kg给药后,用雄性ICR小鼠(每组n = 6,体重20-30g)研究磺胺抑素的药代动力学。对于静脉内给药制剂,将磺胺替尼溶于DMSO(0.25%) - 溶质(10%) - 乙醇(10%) - 生理盐水(79.75%),浓度为0.25mg / mL。并且用0.5%CMC-Na制备po剂量制剂(1mg / mL)。静脉注射或口服给药后,在0(关闭前),5,15,30分钟和1,1.5,2,4,8,24小时通过眼静脉收集血液样品,用肝素-Na凝固。离心后,分离血浆样品,用含有内标的乙腈进行蛋白质沉淀[2]。 |
| 参考文献 |
| 密度 | 1.3±0.1 g/cm3 |
|---|---|
| 沸点 | 712.9±70.0 °C at 760 mmHg |
| 分子式 | C24H28N6O3S |
| 分子量 | 480.582 |
| 闪点 | 385.0±35.7 °C |
| 精确质量 | 480.194366 |
| LogP | 3.06 |
| 外观性状 | 浅黄色粉末 |
| 蒸汽压 | 0.0±2.3 mmHg at 25°C |
| 折射率 | 1.658 |
| 储存条件 | 室温 |