
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:(E)-4-(2-(N-((4-甲氧基苯基)磺酰基)乙酰氨基)苯乙烯基)吡啶1-氧化物
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:HMN-214
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:(E)-4-[2-[2-[N-乙酰基-N-[(4-甲氧基苯基)磺酰]氨基]苯基]乙烯基]吡啶 1-氧化物
- 纯度:98.0%
- 规格:500mg/1g/10mg/10mg
- 联系人:李先生
- 联系电话:18916172912
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:HMN-214
- 纯度:98.0%
- 规格:25mg/5mg/1ml/100mg
- 联系人:阿拉丁
- 联系电话:400-620-6333
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]HMN-214,95%
- 纯度:95.0%
- 规格:25mg
- 联系人:夏言
- 联系电话:18016376636
- 武汉敬康恩生物医药科技有限公司
- 湖北省 武汉市 东西湖区
- 产品名:HMN-214
- 纯度:98.0%
- 规格:1g/5g
- 联系人:周经理
- 联系电话:15871494362
查看所有供应商和价格请点击:
173529-46-9
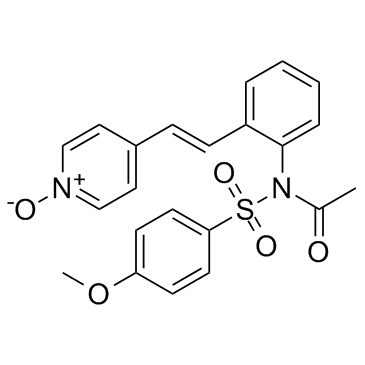
173529-46-9结构式
- 常用中文名:(E)-4-[2-[2-[N-乙酰基-N-[(4-甲氧基苯基)磺酰]氨基]苯基]乙烯基]吡啶 1-氧化物
- 常用英文名:HMN-214
- CAS号:173529-46-9
- 分子式:C22H20N2O5S
- 分子量:424.470
- 相关类别: 生物化工 抑制剂 细胞周期(Cell Cycle) PLK 抑制剂
- 发布时间:2016-02-19 10:02:32
- 更新时间:2024-01-02 11:09:57
-
HMN-214 是一种可口服的 HMN-176 前体药物,为 plk1 抑制剂,具有抗肿瘤活性。
-
一、体外活性:
HMN-214是一种快速转化为HMN-176的口服前药。 HMN-214的体外数据很少。然而,HMN-214的活性代谢物HMN-176对包括HeLa,PC-3,DU-145,MIAPaCa-2,U937,MCF-7,A549和WiDr等多种癌细胞显示出有效和广谱的肿瘤活性,平均IC50为118 nM。250 nM-2.5 μM HMN-176抑制减数分裂纺锤体装配。2.5 μM HMN-176 也抑制人中心体形成微管。这些结果表明HMN-176的抗肿瘤活性至少部分是通过在有丝分裂过程中破坏中心体介导的MT组装来实现的。2.5 μM HMN-176延迟人RPE1和CFPAC-1细胞中合适纺锤体装配检验点的形成。由于NF-Y转录因子与MDR1启动子结合的紊乱,HMN-176(3 μM)下调多药耐药基因(MDR1)的表达。在HeLa细胞中,HMN-176(3 μM)阻断G2/M期的细胞周期。HMN-176对包括P388/CDDP,P388/VCR,K2/CDDP和K2/VP-16的耐药性人和鼠细胞系也具有细胞毒性,IC50值范围为143 nM-265 nM。
二、体内活性:
HMN-214是HMN-176的口服药物前体,具有较好的口服吸收性。在携带多药耐药KB-A.1细胞的裸鼠模型中,HMN-214(10 mg/kg-20 mg/kg)显著抑制MDR1mRNA表达。在PC-3,A549和WiDr细胞的小鼠异种移植模型中,HMN-214(10 mg/kg-20 mg/kg)抑制肿瘤生长。
| 中文名 | (E)-4-[2-[2-[N-乙酰基-N-[(4-甲氧基苯基)磺酰]氨基]苯基]乙烯基]吡啶 1-氧化物 |
|---|---|
| 英文名 | N-(4-methoxyphenyl)sulfonyl-N-[2-[(E)-2-(1-oxidopyridin-1-ium-4-yl)ethenyl]phenyl]acetamide |
| 英文别名 |
Acetamide, N-[(4-methoxyphenyl)sulfonyl]-N-[2-[(E)-2-(1-oxido-4-pyridinyl)ethenyl]phenyl]-
N-[(4-Methoxyphenyl)sulfonyl]-N-{2-[(E)-2-(1-oxido-4-pyridinyl)vinyl]phenyl}acetamide IVX-214 HMN-214 (E)-4-(2-(2-(N-Acetyl-N-((p-methoxyphenyl)sulfonyl)amino)phenyl)ethenyl)pyridine 1-oxide cc-31 Acetamide,N-((4-methoxyphenyl)sulfonyl)-N-(2-((1E)-2-(1-oxido-4-pyridinyl)ethenyl)phenyl) Acetamide,N-((4-methoxyphenyl)sulfonyl)-N-(2-(2-(1-oxido-4-pyridinyl)ethenyl)phenyl)-,(E) |
| 描述 | HMN-214 是一种可口服的 HMN-176 前体药物,为 plk1 抑制剂,具有抗肿瘤活性。 |
|---|---|
| 相关类别 | |
| 靶点 |
PLK1 |
| 体外研究 | HMN-214是HMN-176的前体药物。 HMN-176显示出针对22种人肿瘤细胞系的有效活性,平均IC50为118 nM [1]。 HMN-176(3-300nM)在HeLa细胞中以剂量依赖性方式抑制由MDR1启动子驱动的荧光素酶表达。 HMN-176(30-3000 nM)也剂量依赖性地抑制Y盒上的复合物形成[3]。相对于载体对照,HMN-214(3.3μM)在PC3-PSMA细胞中用1,4C-1,4Bis聚合物(11倍)和PEI(37倍)增强荧光素酶表达。 HMN-214(≥3.3μM)显着降低细胞增殖,导致MB49细胞中细胞形态发生相当大的变化[4]。 |
| 体内研究 | HMN-214(33mg/kg,口服)在大鼠中转化为HMN-176。 HMN-214对动作和胫神经的传导速度和动作电位的幅度没有影响。 HMN-214(20mg/kg,口服)在小鼠中显示出抗肿瘤活性[1]。 HMN-214(10,20 mg/kg,po)降低了携带KB-和KB-A.1。衍生肿瘤的裸鼠中MDR1 mRNA的表达[3]。 |
| 细胞实验 | 使用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物,相对于未处理的对照细胞(处理为100%存活或活对照),在不同处理条件下的细胞增殖进行定量。 (MTT),黄色试剂,通过活细胞转化为甲(紫色染料)。对于筛选实验,转染在96孔细胞培养板中进行,每孔接种50,000个细胞。转染48小时后,向细胞中加入10μLMTT试剂,并在37℃下孵育2-4小时,然后通过加入20μLMTT去污剂裂解细胞,并在室温下再孵育2小时。温度。抑制剂剂量优化转染在24孔板中进行,每孔接种50,000个细胞。 48小时后,加入20μLMTT试剂,然后加入100μLMTT洗涤剂裂解2小时[4]。 |
| 动物实验 | 通过逐渐添加0.5%甲基纤维素4000溶液使地面HMN-214用玛瑙研杵悬浮以制备3mg / mL悬浮液。另外用甲基纤维素4000溶液稀释,得到适当浓度的悬浮液。通过皮下移植到裸鼠中预先培养肿瘤组织。除去所得肿瘤,切成8mm 3的立方体片段,并用套管针将sc移植到裸鼠的右腋窝区域。当肿瘤的理论体积达到约145mm 3时,开始口服HMN-214(第1天)[3]。 |
| 参考文献 |
| 密度 | 1.2±0.1 g/cm3 |
|---|---|
| 沸点 | 663.1±65.0 °C at 760 mmHg |
| 分子式 | C22H20N2O5S |
| 分子量 | 424.470 |
| 闪点 | 354.8±34.3 °C |
| 精确质量 | 424.109283 |
| PSA | 97.52000 |
| LogP | 1.85 |
| 蒸汽压 | 0.0±2.0 mmHg at 25°C |
| 折射率 | 1.598 |
| 储存条件 | -20°C |