857036-77-2
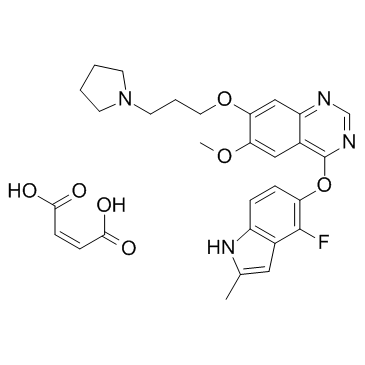
857036-77-2 structure
- Name: Cediranib (maleate)
- Chemical Name: (Z)-but-2-enedioic acid,4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)quinazoline
- CAS Number: 857036-77-2
- Molecular Formula: C29H31FN4O7
- Molecular Weight: 566.57700
- Catalog: Signaling Pathways Autophagy Autophagy
- Create Date: 2018-06-25 17:38:32
- Modify Date: 2024-01-03 13:56:47
-
Cediranib maleate (AZD-2171 maleate) is a highly potent, orally available VEGFR inhibitor with IC50s of <1, <3, 5, 5, 36, 2 nM for Flt1, KDR, Flt4, PDGFRα, PDGFRβ, c-Kit, respectively.
| Name | (Z)-but-2-enedioic acid,4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)quinazoline |
|---|---|
| Synonyms |
AZD2171 maleate
Cediranib maleate (JAN/USAN) UNII-68AYS9A614 Cediranib maleate [USAN:JAN] [14C]-Cediranib maleate 4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazoline maleate Cediranib maleate Cediranib maleate Cediranib (maleate) |
| Description | Cediranib maleate (AZD-2171 maleate) is a highly potent, orally available VEGFR inhibitor with IC50s of <1, <3, 5, 5, 36, 2 nM for Flt1, KDR, Flt4, PDGFRα, PDGFRβ, c-Kit, respectively. |
|---|---|
| Related Catalog | |
| Target |
Flt-1:5 nM (IC50) KDR:1 nM (IC50) Flt-4:3 nM (IC50) PDGFRα:36 nM (IC50) PDGFRβ:5 nM (IC50) c-Kit:2 nM (IC50) |
| In Vitro | In human umbilical vein endothelial cells, Cediranib inhibits VEGF-stimulated proliferation and KDR phosphorylation with IC50 values of 0.4 and 0.5 nM, respectively. In a fibroblast/endothelial cell coculture model of vessel sprouting, Cediranib also reduces vessel area, length, and branching at subnanomolar concentrations[1]. |
| In Vivo | Once-daily oral administration of Cediranib ablates experimental (VEGF-induced) angiogenesis and inhibits endochondral ossification in bone or corpora luteal development in ovary; physiologic processes that are highly dependent upon neovascularization. The growth of established human tumor xenografts (colon, lung, prostate, breast, and ovary) in athymic mice is inhibited dose-dependently by Cediranib, with chronic administration of 1.5 mg per kg per day producing statistically significant inhibition in all models. A histologic analysis of Calu-6 lung tumors treated with Cediranib reveals a reduction in microvessel density within 52 hours that becomes progressively greater with the duration of treatment. These changes are indicative of vascular regression within tumors[1]. |
| Kinase Assay | The inhibitory activity of Cediranib is determined against a range of recombinant tyrosine kinases [KDR, Flt-1, Flt-4, c-Kit, PDGFR-α, PDGFR-β, CSF-1R, Flt-3, FGFR1, Src, Abl, epidermal growth factor receptor (EGFR), ErbB2, Aur-A, and Aur-B] using ELISA methodology[1]. |
| Cell Assay | Proliferation of MG63 osteosarcoma cells is induced by PDGF-AA, which selectively activates PDGFR-α homodimer signaling. Cells are cultured in DMEM without phenol red containing 1% charcoal stripped FCS, 2 mM glutamine, and 1% nonessential amino acids for 24 hours. Cediranib or vehicle is added with PDGF-AA ligand (50 ng/mL) and plates reincubated for 72 hours. Cellular proliferation is determined using a bromodeoxyuridine[1]. |
| Animal Admin | Rats: Young female Alderley Park rats (6 weeks of age, Wistar derived, n=5) are dosed orally, once daily for 28 days with Cediranib (1.25-5 mg per kg per day) or vehicle. Additional rats (five per group) are treated with Cediranib (5 mg per kg per day) or vehicle for 28 days and maintained for a further 28 days without treatment, to examine the effect of compound withdrawal. Histologic paraffin wax sections of the femorotibial joints and ovaries are stained with H&E. Morphometric image analysis of femorotibial sections is done, with growth plate areas from both the femur and tibia in each joint being combined for an analysis of the effect of compound treatment. The area of corpora lutea in H&E-stained ovary sections is similarly determined by morphometric analysis[1]. |
| References |
| Molecular Formula | C29H31FN4O7 |
|---|---|
| Molecular Weight | 566.57700 |
| Exact Mass | 566.21800 |
| PSA | 147.10000 |
| LogP | 4.87390 |
| Storage condition | -20℃ |