905854-02-6
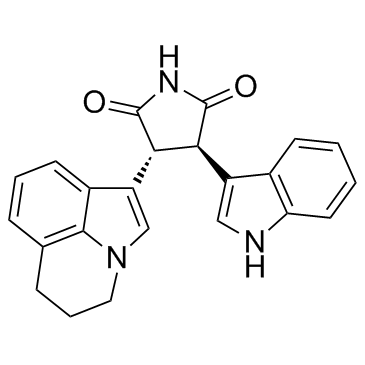
905854-02-6 structure
- Name: Tivantinib (ARQ 197)
- Chemical Name: 3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)-pyrrolidine-2,5-dione
- CAS Number: 905854-02-6
- Molecular Formula: C23H19N3O2
- Molecular Weight: 369.416
- Catalog: Biochemical Inhibitor Protein tyrosine kinase
- Create Date: 2018-12-10 19:34:05
- Modify Date: 2024-01-02 20:49:42
-
Tivantinib is a novel and highly selective c-Met tyrosine kinase inhibitor with Ki of 355 nM.
| Name | 3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)-pyrrolidine-2,5-dione |
|---|---|
| Synonyms |
(3R,4R)-3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione
(-)-3(R),4(R)-3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione (3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)-2,5-pyrrolidinedione (3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]chinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidin-2,5-dion ARQ 197 Tivantinib (ARQ 197) Tivantinib/ARQ197/ARQ-197 2,5-Pyrrolidinedione, 3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)-, (3R,4R)- (-)-trans-3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione Tivantinib ARQ-197 (-)-3(R),4(R)-3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione,(-)-trans-3-(5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione ARQ197 |
| Description | Tivantinib is a novel and highly selective c-Met tyrosine kinase inhibitor with Ki of 355 nM. |
|---|---|
| Related Catalog | |
| Target |
Ki: 355 nM (c-Met)[1] |
| In Vitro | Tivantinib (ARQ 197) selectively inhibits c-Met activity in cell-free and cell-based assays. c-Met-expressing cancer cell lines treated with Tivantinib display either a dose-dependent loss of proliferative capacity or caspase-dependent apoptosis that positively correlates with either ligand-dependent c-Met activity or constitutively active c-Met. To examine the biochemical mode of inhibition of Tivantinib, kinetic analyses are done using recombinant human c-Met in a filtermat-based assay. The Km of ATP is 50.5±2.2 μM, which is similar to the Km value of ATP. In these kinetic studies, Tivantinib inhibits human recombinant c-Met with a calculated inhibitory constant (Ki) of ~355 nM. In vitro exposure to Tivantinib inhibits constitutive c-Met phosphorylation in HT29 and MKN-45 cells, and HGF-induced c-Met phosphorylation in MDA-MB-231 and NCI-H441 cells with an IC50 of 100 to 300 nM[1]. Tivantinib is a low-molecular-weight compound, and is the first in class orally available selective inhibitor of c-Met[2]. |
| In Vivo | Pharmacodynamically, the phosphorylation of c-Met in human colon xenograft tumors (HT29) is strongly inhibited by Tivantinib (ARQ 197), as assessed by a dramatic reduction of c-Met autophosphorylation 24 hours after a single oral dose of 200 mg/kg of Tivantinib. This same dosage in mice shows that tumor xenografts are exposed to sustained plasma levels of Tivantinib, consistent with the observed pharmacodynamic inhibition of c-Met phosphorylation and inhibition of proliferation of c-Met harboring cancer cell lines. A Cmax of 5.73 μg/mL (13 μM), an area under the concentration-time curve of 12.1 μg/mL h, and a t1/2 of 2.4 hours are measured. Plasma levels of Tivantinib 10 hours after dosing are determined to be 1.3 μM, >3-fold above the biochemical inhibitory constant of Tivantinib for c-Met[1]. |
| Kinase Assay | Recombinant c-Met protein (50 ng) comprising residues 974 to 1390 is incubated in a reaction buffer [50 mM Tris-HCl (pH 7.5), 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2, 1 mM EGTA, 0.02 mg/mL bovine serum albumin, and 10% glycerol] with various concentrations of Tivantinib or DMSO in a total volume of 20 μL. After incubating for 20 minutes at room temperature, 20 μL of 20 μM poly-Glu-Tyr substrate and 20 μL of increasing amounts of cold ATP containing 1.5 μCi of [γ-33P]ATP are added to initiate the reaction. The reaction is stopped after 5, 10, 20, 40, and 60 minutes by the addition of 10% phosphoric acid and 10 μL aliquots of the reaction mixture are spotted onto P30 filtermat in triplicate. The filters are washed thrice for 5 minutes with 0.75% phosphoric acid and once with methanol for 2 minutes. The level of radioactivity is determined by using a Wallac TriLux MicroBeta liquid scintillation counter. Nonspecific binding is determined by conducting the assay in the absence of enzyme and then subtracting the value from each of the experimental values. Reaction rates are determined in the linear range of each reaction and GraphPad Prism software is used to calculate the Km and Vmax values[1]. |
| Cell Assay | HT29, MKN-45, MDA-MB-231, and NCI-H441 (lung cancer) human cancer cells are seeded in 96-well plates overnight in a medium with 10% FBS. Each cell line is optimized for seeding cell number to ensure a similar degree of confluence at the end of the experiment in nontreated (control) wells. The next day, cells are treated with different concentrations of Tivantinib for 24 hours at 37°C. After ARQ 197 treatment, the drug-containing medium is removed, and cells are washed twice with PBS and incubated in a drug-free medium for an additional 48 hours. Cells are then incubated and stained for 4 hours with the MTS reagent (final concentration of 0.5 mg/mL) per well and are lysed. The results are quantitated by spectrophotometry at λ=450 nm[1]. |
| Animal Admin | Mice[1] Female athymic nude mice are acclimated to the animal housing facility for at least 1 week before the study. Efficacy studies are done in athymic mice bearing HT29, MKN-45, or MDA-MB-231 tumor xenografts to determine the effect of ARQ 197 on tumor growth. Tumor cells [5×106 (HT29) and 8×106 (MKN-45 and MDA-MB-231) cells/animal] are inoculated s.c. on day 0. Tumor dimensions are measured by a digital caliper and tumor volumes are calculated as length×width2/2. When tumors reached a volume of ~100 mm3, mice are randomized into groups and treated daily with orally administered vehicle control or 200 mg/kg Tivantinib formulated in polyethylene glycol 400/20% Vitamin E tocopheryl polyethylene glycol succinate (60:40) at 30 mg/mL, for 5 consecutive days, followed by a 2-day dosing holiday for four cycles. Therefore, each animal received a total of 20 doses. Results are expressed as mean tumor volume±SEM. To assess differences in tumor size between groups, a Mann-Whitney nonparametric t test is performed and significance is defined as P<0.05. Rats[2] Six male Sprague-Dawley rats (180-220 g) are used to study the pharmacokinetics of Tivantinib. Diet is prohibited for 12 h before the experiment but water is freely available. Blood samples (0.3 mL) are collected from the tail vein into heparinized 1.5 mL polythene tubes at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h after oral administration of Tivantinib (10 mg/kg). The samples are immediately centrifuged at 4000g for 8 min. The plasma obtained (100 μL) is stored at -20°C until analysis. Plasma Tivantinib concentration versus time data for each rat is analyzed by DAS (Drug and statistics) software. |
| References |
| Density | 1.5±0.1 g/cm3 |
|---|---|
| Boiling Point | 716.0±60.0 °C at 760 mmHg |
| Molecular Formula | C23H19N3O2 |
| Molecular Weight | 369.416 |
| Flash Point | 386.8±32.9 °C |
| Exact Mass | 369.147736 |
| PSA | 66.89000 |
| LogP | 3.26 |
| Vapour Pressure | 0.0±2.3 mmHg at 25°C |
| Index of Refraction | 1.797 |
| Storage condition | -20℃ |
|
~%
905854-02-6 |
| Literature: US2011/160242 A1, ; Page/Page column 27-29 ; |
|
~%
905854-02-6 |
| Literature: US2011/160242 A1, ; |
|
~%
905854-02-6 |
| Literature: US2011/160242 A1, ; |
|
~%
905854-02-6 |
| Literature: US2011/160242 A1, ; |
|
~%
905854-02-6 |
| Literature: US2011/160242 A1, ; |


| Precursor 4 | |
|---|---|
| DownStream 0 | |