
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:GSK-J4
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:GSK J4 HCl
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:GSK J4 HCl(free base)
- 纯度:98.0%
- 规格:10mg/1g/50mg/1g
- 联系人:李先生
- 联系电话:18916172912
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:GSK-J4
- 纯度:98.0%
- 规格:10mg/25mg/5mg/1ml
- 联系人:阿拉丁
- 联系电话:400-620-6333
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]GSK J4 HCl,99%
- 纯度:99.0%
- 规格:1mg/10mg/1ml/50mg
- 联系人:夏言
- 联系电话:18016376636
- 广东翁江化学试剂有限公司
- 广东省 韶关市 翁源县
- 产品名:N-[2-(2-吡啶基)-6-(1,2,4,5-四氢-3H-3-苯并氮杂卓-3-基)-4-嘧啶基]-BETA-丙氨酸乙酯
- 纯度:98.0%
- 规格:5mg
- 联系人:朱丽丹
- 联系电话:13927877242
- 武汉壹加壹生物科技有限公司
- 湖北省 武汉市 江夏区
- 产品名:N-[2-(2-吡啶基)-6-(1,2,4,5-四氢-3H-3-苯并氮杂卓-3-基)-4-嘧啶基]-BETA-丙氨酸乙酯
- 纯度:98.0%
- 规格:1g
- 联系人:刘经理
- 联系电话:16602739303
查看所有供应商和价格请点击:
1373423-53-0
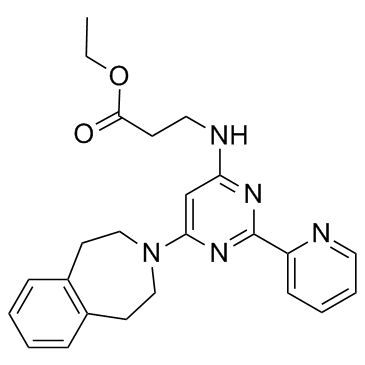
1373423-53-0结构式
- 常用中文名:GSK-J4
- 常用英文名:GSK-J4
- CAS号:1373423-53-0
- 分子式:C24H27N5O2
- 分子量:417.504
- 相关类别: 生物化工 抑制剂 表观遗传学(Epigenetics) Histone Demethylase 抑制剂
- 发布时间:2017-03-29 10:35:50
- 更新时间:2024-01-02 16:22:00
-
GSK-J4是一种有效的H3K27me3组蛋白赖氨酸脱甲基酶 (KDM) 抑制剂,抑制 KDM6B和KDM6A的 IC50 分别为8.6 μM 和 6.6 μM。
| 中文名 | N-[2-(2-吡啶基)-6-(1,2,4,5-四氢-3H-3-苯并氮杂卓-3-基)-4-嘧啶基]-BETA-丙氨酸乙酯 |
|---|---|
| 英文名 | ethyl 3-[[2-pyridin-2-yl-6-(1,2,4,5-tetrahydro-3-benzazepin-3-yl)pyrimidin-4-yl]amino]propanoate |
| 英文别名 |
GSK-J4
Ethyl N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl)-4-pyrimidinyl]-β-alaninate GSK J4 HCl β-Alanine, N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl)-4-pyrimidinyl]-, ethyl ester |
| 描述 | GSK-J4是一种有效的H3K27me3组蛋白赖氨酸脱甲基酶 (KDM) 抑制剂,抑制 KDM6B和KDM6A的 IC50 分别为8.6 μM 和 6.6 μM。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: 8.6 µM (KDM6B), 6.6 µM (KDM6A)[5] |
| 体外研究 | GSK-J4在Flag-JMJD3转染的HeLa细胞中具有细胞活性,其中GSK-J4阻止JMJD3诱导的核H3K27me3免疫染色的丧失。施用GSK-J4增加未转染细胞中的总核H3K27me3水平。 GSK-J4显着降低了34种LPS驱动的细胞因子中16种的表达,包括肿瘤坏死因子-α(TNF-α)[1]。 GSK-J4(10,25 nM)作用于促进Treg细胞分化,改善Treg稳定性和抑制能力的DC,而不影响Th1和Th17细胞的分化[2]。 GSK-J4抑制KK6家族的H3K27me3去甲基化酶JMJD3和UTX。 GSK-J4抑制由TGF-β1诱导的JMJD3表达[3]。 GSK-J4抑制雌性胚胎干细胞中Xist,Nodal和HoxC13的H3K4去甲基化[4]。 |
| 体内研究 | GSK-J4(0.5 mg/kg,ip)可显着降低实验性自身免疫性脑脊髓炎小鼠模型的严重程度并延缓其发病[2]。 |
| 动物实验 | 6至8周龄的雌性C57BL / 6 WT小鼠通过皮下注射(sc)注射50μg髓鞘少突胶质细胞糖蛋白35-55肽(pMOG),其在完全弗氏佐剂(CFA)中乳化,补充有热灭活的结核分枝杆菌H37 RA。此外,小鼠在第0天和第2天接受500ng百日咳毒素的腹膜内注射(ip)。根据以下评分标准每天评估临床体征:0,没有可检测的迹象; 1,松弛的尾巴; 2,后肢无力或步态异常; 3,完全后肢麻痹; 4,前肢和后肢麻痹; 5,垂死或死亡。在二甲基亚砜(DMSO)中制备GSK-J4的储备溶液42mg / mL(100mM)以保持稳定性。在注射之前,将储备溶液用乙醇(DMSO:乙醇,1:10v / v)稀释1/10并在PBS中达到140μg/ mL的终浓度。在全身药物评估实验中,每只小鼠每天接受100μL含有14.0μgGSK-J4(相当于0.56mg / kg药物)的该溶液的ip注射(从第0-5天开始)。对照小鼠在同一时期接受100μL载体。在其他EAE实验中,来自WT小鼠的106个骨髓来源的DC用GSK-J4或单独的载体处理16小时,用5μg/ mL的pMOG脉冲4小时,然后静脉内转移到WT C57BL / 6受体小鼠14中。和EAE诱导前7天。在其他过继转移EAE实验中,在25nM GSK-J4存在或不存在下产生的CD4 + Foxp3 + Treg细胞通过细胞分选纯化,然后在EAE诱导前1天将0.75×106静脉内转移到WT C57BL / 6受体小鼠中。 |
| 参考文献 |
[5]. Heinemann B, et al. Inhibition of demethylases by GSK-J1/J4. Nature. 2014 Oct 2;514(7520):E1-2 |
| 密度 | 1.2±0.1 g/cm3 |
|---|---|
| 沸点 | 581.2±50.0 °C at 760 mmHg |
| 分子式 | C24H27N5O2 |
| 分子量 | 417.504 |
| 闪点 | 305.3±30.1 °C |
| 精确质量 | 417.216461 |
| PSA | 80.24000 |
| LogP | 3.75 |
| 外观性状 | white to beige |
| 蒸汽压 | 0.0±1.6 mmHg at 25°C |
| 折射率 | 1.615 |
| 储存条件 | 2-8°C |
| 水溶解性 | DMSO: soluble20mg/mL, clear |