TTA-A2
更新时间:2024-07-18 12:08:23
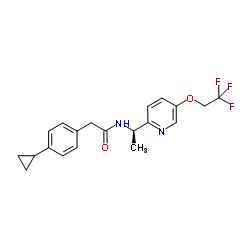
TTA-A2结构式

|
常用名 | TTA-A2 | 英文名 | TTA-A2 |
|---|---|---|---|---|
| CAS号 | 953778-63-7 | 分子量 | 378.388 | |
| 密度 | 1.3±0.1 g/cm3 | 沸点 | 538.5±50.0 °C at 760 mmHg | |
| 分子式 | C20H21F3N2O2 | 熔点 | N/A | |
| MSDS | N/A | 闪点 | 279.5±30.1 °C |
TTA-A2用途TTA-A2 是一种强效、选择性和口服活性的 T 型电压门控钙通道 (calcium channel) 拮抗剂,可减少孕烷 X 受体 (PXR) 的激活。TTA-A2 对 Cav3.1 (a1G) 和 Cav3.2 (a1H) 通道在 -80 mV 和- 100 mV 保持电位上具有同样的作用,IC50 值分别为 89 nM 和 92 nM。TTA-A2 可用于多种人类神经系统疾病的研究,包括睡眠障碍和癫痫。 |

| 英文名 | TTA-A2 |
|---|---|
| 英文别名 | 更多 |
| 描述 | TTA-A2 是一种强效、选择性和口服活性的 T 型电压门控钙通道 (calcium channel) 拮抗剂,可减少孕烷 X 受体 (PXR) 的激活。TTA-A2 对 Cav3.1 (a1G) 和 Cav3.2 (a1H) 通道在 -80 mV 和- 100 mV 保持电位上具有同样的作用,IC50 值分别为 89 nM 和 92 nM。TTA-A2 可用于多种人类神经系统疾病的研究,包括睡眠障碍和癫痫。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: 98 nM ( α1I at membrane holding potentials of -80 mV) IC50: 3.7 μM (α1I at membrane holding potentials of -100 mV)[1] |
| 体外研究 | 在标准电压钳电生理实验中,TTA-A2对α1I具有状态依赖性的抑制作用,在膜保持电位为-80和-100mv时,其效价分别为98nm和3.7μM。它还对Cav1.2(L型)、Cav2.1(P/Q型)、Cav2.2(N型)和Cav2.3(R型)通道具有良好的选择性,这些通道在80 mV时的IC50值都大于30μM[1]。TTA-A2在α1I结合实验中表现出很高的亲和力,Ki为1.2nm,对hERG钾通道和L型钙通道(IC50>10μM)具有良好的选择性[1]。 |
| 体内研究 | TTA-A2(口服灌胃;3mg/kg;单剂量)对大鼠睡眠结构有显著影响。给药后即刻活动性觉醒减少,同时增加delta睡眠和减少REM睡眠。此外,在大鼠中,这些效应在给药后持续4小时[1]。TTA-A2(口服灌胃;10mg/kg;每日一次;5天)对反复丘脑皮质神经网络活动有选择性作用,在野生型小鼠中抑制主动觉醒和促进慢波睡眠,但在缺乏Cav3.1和Cav3.3的小鼠中没有[2]。动物模型:野生型和双Cav3.1/Cav3.3基因敲除C57BL6/Sv129背景小鼠[2]剂量:10mg/kg给药:经口灌胃;10mg/kg;每日1次;5天结果:野生型小鼠活动觉醒受阻,促进慢波睡眠,而突变型小鼠没有。 |
| 参考文献 |
[1]. Thomas S Reger, et al.Pyridyl amides as potent inhibitors of T-type |
| 密度 | 1.3±0.1 g/cm3 |
|---|---|
| 沸点 | 538.5±50.0 °C at 760 mmHg |
| 分子式 | C20H21F3N2O2 |
| 分子量 | 378.388 |
| 闪点 | 279.5±30.1 °C |
| 精确质量 | 378.155518 |
| LogP | 3.58 |
| 蒸汽压 | 0.0±1.4 mmHg at 25°C |
| 折射率 | 1.545 |
| 2-(4-Cyclopropylphenyl)-N-{(1R)-1-[5-(2,2,2-trifluoroethoxy)-2-pyridinyl]ethyl}acetamide |
| TTA-A2 |
| Benzeneacetamide, 4-cyclopropyl-N-[(1R)-1-[5-(2,2,2-trifluoroethoxy)-2-pyridinyl]ethyl]- |

- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:TTA-A2
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:TTA-A2
- 产品网址:https://www.med-bio.cn/product/45766.html
- 纯度:98.0%
- 规格:1mg
- 联系人:李先生
- 联系电话:18916172912
- 武汉敬康恩生物医药科技有限公司
- 湖北省 武汉市 东西湖区
- 产品名:"(R)-2-(4-cyclopropylphenyl)-N-(1-(5-(2,2,2-trifluoroethoxy)pyridin-2-yl)ethyl)acetamide "
- 纯度:98.0%
- 规格:1g
- 联系人:周经理
- 联系电话:15871494362
查看所有供应商和价格请点击: