923032-37-5
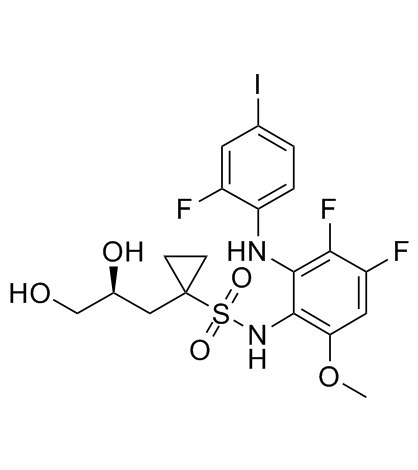
923032-37-5 structure
- Name: Refametinib
- Chemical Name: N-[3,4-difluoro-2-(2-fluoro-4-iodoanilino)-6-methoxyphenyl]-1-[(2S)-2,3-dihydroxypropyl]cyclopropane-1-sulfonamide
- CAS Number: 923032-37-5
- Molecular Formula: C19H20F3IN2O5S
- Molecular Weight: 572.337
- Catalog: Biochemical Inhibitor Mitogen-activated protein kinase (MAPK) MEK inhibitor
- Create Date: 2018-07-07 16:26:16
- Modify Date: 2024-01-02 14:28:35
-
Refametinib is a potent, selective, allosteric MEK1/MEK2 inhibitor with IC50s of 19 nM and 47 nM, respectively.
| Name | N-[3,4-difluoro-2-(2-fluoro-4-iodoanilino)-6-methoxyphenyl]-1-[(2S)-2,3-dihydroxypropyl]cyclopropane-1-sulfonamide |
|---|---|
| Synonyms |
3e8n
BAY 86-9766 UNII-JPX07AFM0N N-{3,4-Difluoro-2-[(2-fluoro-4-iodophenyl)amino]-6-methoxyphenyl}-1-[(2S)-2,3-dihydroxypropyl]cyclopropanesulfonamide Cyclopropanesulfonamide, N-[3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]-6-methoxyphenyl]-1-[(2S)-2,3-dihydroxypropyl]- RDEA119 Refametinib |
| Description | Refametinib is a potent, selective, allosteric MEK1/MEK2 inhibitor with IC50s of 19 nM and 47 nM, respectively. |
|---|---|
| Related Catalog | |
| Target |
MEK1:19 nM (IC50) MEK2:47 nM (IC50) |
| In Vitro | Refametinib (RDEA119, BAY 869766) selectively binds directly to an allosteric pocket in the MEK1/2 enzymes. Refametinib potently inhibits MEK activity in enzyme inhibition assays in a non-ATP-competitive manner (MEK1 IC50=19 nM, MEK2 IC50=47 nM) determined through incorporation of radioactive phosphate from ATP into ERK as substrate. Refametinib potently inhibits MEK activity as measured by phosphorylation of ERK1/2 across several human cancer cell lines of different tissue origins and BRAF mutational status with EC50 values ranging from 2.5 to 15.8 nM. Refametinib inhibits anchorage-dependent growth of human cancer cell lines harboring the gain-of-function V600E BRAF mutant with GI50 values ranging from 67 to 89 nM. In contrast, Refametinib has significantly less growth-inhibitory potency against cell lines with wild-type BRAF (A431 cells) or MDA-MB-231 cells harboring a BRAF mutation G464V that shows minimal (<2-fold increase) enhancement of inherent kinase activity. Under anchorage-independent conditions, GI50 values for all cell lines tested are similar (40-84 nM). MDA-MB-231 and A431 cells are significantly more sensitive to Refametinib under anchorage-independent conditions[1]. |
| In Vivo | Refametinib (RDEA119, BAY 869766) is an orally available, potent, non-ATP-competitive, highly selective inhibitor of MEK1/2, which is active in human tumor xenograft models and is well tolerated within the therapeutic exposure range in animals.The human melanoma A375 tumor xenograft is found to be sensitive to Refametinib treatment with 54% and 68% tumor growth inhibition (TGI) seen with 25 and 50 mg/kg/d administered orally on a once daily ×14 schedule. Significant tumor growth delay (TGD) and regressions are also observed in A375 tumors on this once-daily schedule. For example, five to eight complete or partial responses (CR/PR) and up to six tumor-free survivors (TFS) are observed. Administering Refametinib every other day at 100 mg/kg is less effective than daily dosing with either 25 or 50 mg/kg. When Refametinib is dosed on a twice-daily schedule, it is more effective than once-daily schedules[1]. |
| Kinase Assay | MEK1 kinase activity is determined using mERK2 K52A T183A as the substrate. Recombinant MEK1 enzyme (5 nM) is first activated by 0.02 unit or 1.5 nM of RAF1 in the presence of 25 mM HEPES (pH 7.8), 1 mM MgCl2, 50 mM NaCl, 0.2 mM EDTA, and 50 μM ATP for 30 min at 25°C. The reactions are initiated by adding 2 μM of mERK2 K52A T183A and 2.5 μCi [γ-33P]ATP in a total volume of 20 μL. The MEK2 kinase activity is determined similarly except that activation by RAF1 is not needed and 11 nM of MEK2 enzyme (active) are used in the assays. Kinase profiling is performed. The Z'-LYTE biochemical assay is used. Refametinib is assayed in quadruplicate at 10 μM against 205 kinases[1]. |
| Cell Assay | For anchorage-dependent growth inhibition experiments, A375, SK-Mel-28, A431, Colo205, HT-29, MDA-MB-231, and BxPC3 cell lines are plated in white 384-well plates at 1,000/20 μL/well or white 96-well microplates at 4,000/100 μL/well. After 24-h incubation at 37°C, 5% CO2, and 100% humidity, Refametinib is incubated for 48 h at 37°C and assayed using CellTiter-Glo. For the 96-well anchorage-independent growth assay, wells of an “ultralow binding” plate are filled with 60 μL of a 0.15% agarose solution in complete RPMI 1640. Then, 60 μL of complete RPMI 1640 containing 9,000 cells in 0.15% agarose are added per well. After 24 h, 60 μL of a 3× drug solution in agarose-free complete RPMI 1640 are added. After 7 d, 36 μL of 6× MTS reagent are added per well. After 2 h at 37°C, absorbance at 490 nm is determined on the M5 plate reader. Nonlinear curve fitting is performed using GraphPad Prism 4[1]. |
| Animal Admin | Mice[1] Female athymic nude mice are used for all the efficacy studies except for the Colo205 study 2, which used male mice. Mice are injected s.c. in the flank with 1×106 tumor cells (Colo205 and A431) or ~1 mm3 tumor fragments (A375 and HT-29). Tumor volumes are monitored by caliper measurements. For efficacy analysis, treatment is initiated when tumors are 80 to 185 mm3. Refametinib (25 and 50 mg/kg/d) is administered by oral gavage once daily or twice daily for 14 d or once every 2 d for 14 doses[1]. |
| References |
| Density | 1.8±0.1 g/cm3 |
|---|---|
| Boiling Point | 566.9±60.0 °C at 760 mmHg |
| Molecular Formula | C19H20F3IN2O5S |
| Molecular Weight | 572.337 |
| Flash Point | 296.7±32.9 °C |
| Exact Mass | 572.008972 |
| PSA | 116.27000 |
| LogP | 4.78 |
| Vapour Pressure | 0.0±1.6 mmHg at 25°C |
| Index of Refraction | 1.660 |
| Storage condition | -20℃ |