1859053-21-6
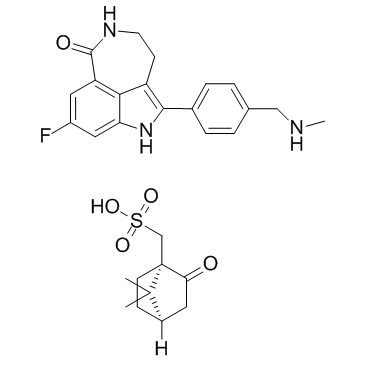
1859053-21-6 structure
- Name: Rucaparib camsylate
- Chemical Name: Rucaparib camsylate
- CAS Number: 1859053-21-6
- Molecular Formula: C29H34FN3O5S
- Molecular Weight: 555.661
- Catalog: Signaling Pathways Cell Cycle/DNA Damage PARP
- Create Date: 2018-03-05 01:23:41
- Modify Date: 2024-01-03 20:25:33
-
Rucaparib is an inhibitor of PARP with a Ki of 1.4 nM for PARP1, and also shows binding affinity to eight other PARP domains.
| Name | Rucaparib camsylate |
|---|---|
| Synonyms |
Bicyclo[2.2.1]heptane-1-methanesulfonic acid, 7,7-dimethyl-2-oxo-, (1S,4R)-, compd. with 8-fluoro-1,3,4,5-tetrahydro-2-[4-[(methylamino)methyl]phenyl]-6H-azepino[5,4,3-cd]indol-6-one (1:1)
UNII:41AX9SJ8KO Rucaparib camsylate CO-338 PF-1367338-BW [(1S,4R)-7,7-Dimethyl-2-oxobicyclo[2.2.1]hept-1-yl]methanesulfonic acid - 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one (1:1) Rucaparib (Camsylate) |
| Description | Rucaparib is an inhibitor of PARP with a Ki of 1.4 nM for PARP1, and also shows binding affinity to eight other PARP domains. |
|---|---|
| Related Catalog | |
| Target |
PARP-1:1.4 nM (Ki) |
| In Vitro | Rucaparib is the most potent PARP inhibitor in enzyme assays (Ki, 1.4 nM), and a possible N-demethylation metabolite of AG14644[1]. The radio-sensitization by Rucaparib is due to downstream inhibition of activation of NF-κB, and is independent of SSB repair inhibition. Rucaparib could target NF-κB activated by DNA damage and overcome toxicity observed with classical NF-κB inhibitors without compromising other vital inflammatory functions[2]. Rucaparib inhibits PARP-1 activity by 97.1% at a concentration of 1 μM in permeabilised D283Med cells[3]. |
| In Vivo | Rucaparib and AG14584 significantly (P < 0.05) increases temozolomide toxicity. Rucaparib (1 mg/kg) significantly increases temozolomide-induced body weight loss. Rucaparib (0.1 mg/kg) results in a 50% increase in the temozolomide-induced tumor growth delay[1]. Rucaparib is not toxic but significantly enhances temozolomide-induced TGD in the DNA repair protein-competent D384Med xenografts. Pharmacokinetics studies also show that Rucaparib is detected in the brain tissue, which indicates that Rucaparib has potential in intra-cranial malignancy therapy[3]. Rucaparib significantly potentiates the cytotoxicity of topotecan and temozolomide in NB-1691, SH-SY-5Y, and SKNBE (2c) cells. Rucaparib enhances the antitumor activity of temozolomide and indicates complete and sustained tumor regression in NB1691 and SHSY5Y xenografts[4]. |
| Kinase Assay | Inhibition of PARP activity in 5×103 D283Med cells is measured using various concentrations of Rucaparib (0-1 μM), compared with DMSO-only. Maximally stimulated PARP activity is measured in samples of permeabilised cells by immunologica[1]. |
| Cell Assay | Medulloblastoma cell lines are seeded in 96-well plates at a density of 1×103, 3×103 and 3×103, respectively. At 24 hours (D384Med) or 48 hours (D283Med and D425Med) after seeding, the cells are exposed to various concentrations of temozolomide in the presence or absence of 0.4 μM Rucaparib. After 3 days (D425Med and D384Med) or 5 days (D283Med) of culture, cell viability is evaluated by a XTT cell proliferation kit assay. Cell growth is expressed as a percentage in relation to DMSO or 0.4 μM Rucaparib-alone controls. The concentration of temozolomide, alone or in combination with Rucaparib that inhibited growth by 50% (GI50) is calculated. The potentiation factor 50 (PF50) is defined as the ratio of the GI50 of temozolomide in the presence of Rucaparib to the GI50 of temozolomide alone[1]. |
| Animal Admin | A single dose of temozolomide is administrated p.o. as a suspension in saline at 200 mg/kg either alone or in combination with a single i.p. administration of PARP inhibitor administered at 0.1 [Rucaparib and MS-AG14644 (equivalent to 0.078 mg/kg free AG14644 only)], 1.0, and 10 mg/kg (for the mesylate salts equivalent to 0.79 and 7.9 mg/kg free AG14451 and AG14452 and 0.78 and 7.8 free AG14531 and AG14644). Control animals are treated with either normal saline p.o. and i.p or normal saline p.o and PARP inhibitor 10 mg/kg i.p[1]. |
| References |
| Molecular Formula | C29H34FN3O5S |
|---|---|
| Molecular Weight | 555.661 |
| Exact Mass | 555.220337 |
| Storage condition | 2-8℃ |