
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:PF-04457845
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:13311869306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:PF-04457845
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:PF-04457845
- 纯度:98.0%
- 规格:10mg
- 联系人:李先生
- 联系电话:18916172912
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:PF-04457845
- 纯度:98.0%
- 规格:10mg/5mg/50mg/1g
- 联系人:阿拉丁
- 联系电话:400-620-6333
- 上海创赛科技有限公司
- 上海市 上海市 嘉定区
- 产品名:[Perfemiker]PF-04457845,≥98%
- 纯度:98.0%
- 规格:5mg/10mg
- 联系人:夏言
- 联系电话:18016376636
查看所有供应商和价格请点击:
1020315-31-4

1020315-31-4结构式
- 常用中文名:N-哒嗪-3-基-4-(3-{[5-(三氟甲基)吡啶-2-基]醚}苯亚甲基丙酮)哌啶-1-羧酰胺
- 常用英文名:PF-04457845
- CAS号:1020315-31-4
- 分子式:C23H20F3N5O2
- 分子量:455.43200
- 相关类别: 信号通路 代谢酶/蛋白酶 FAAH
- 发布时间:2016-03-16 08:03:55
- 更新时间:2024-01-08 13:03:44
-
PF-04457845 是一种高效选择性的 FAAH 抑制剂,作用于 hFAAH 和 rFAAH,IC50 分别为 7.2±0.63 nM 和 7.4±0.62 nM。
| 中文名 | N-哒嗪-3-基-4-(3-{[5-(三氟甲基)吡啶-2-基]醚}苯亚甲基丙酮)哌啶-1-羧酰胺 |
|---|---|
| 英文名 | N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide |
| 英文别名 |
CS-0868
N-pyridazin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-yl]oxy}benzylidene)piperidine-1-carboxamide UNII-H4C81M8YYW PF-04457845 |
| 描述 | PF-04457845 是一种高效选择性的 FAAH 抑制剂,作用于 hFAAH 和 rFAAH,IC50 分别为 7.2±0.63 nM 和 7.4±0.62 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: 7.2±0.63 nM (hFAAH), 7.4±0.62 nM (rFAAH)[1] |
| 体外研究 | PF-04457845通过共价,不可逆机制抑制FAAH,其涉及FAAH活性位点丝氨酸亲核体的氨基甲酰化,具有高体外效力(人体FAAH的kinact/Ki和IC50值分别为40300 M-1s-1和7.2 nM) )。相对于丝氨酸水解酶超家族的其他成员,PF-04457845对FAAH具有精确的选择性,如通过基于竞争活性的蛋白质谱分析所证明的。 PF-04457845在10和100μM时完全抑制人和小鼠膜蛋白质组中的FAAH,没有脱靶[1]。 PF-04457845对FAAH具有完全选择性,即使在100μM时,PF-04457845也不会抑制测试组织中的其他FP反应性丝氨酸水解酶[2]。 |
| 体内研究 | 口服施用0.1mg/kg的PF-04457845导致在炎性疼痛的大鼠模型中与10mg/kg的萘普生相当的效力。 PF-04457845的口服给药导致4小时后测量的机械性异常性疼痛的显着抑制,最小有效剂量(MED)为0.1mg/kg。此外,在0.1mg/kg(po)时,PF-04457845抑制疼痛反应的程度与10mg/kg的非甾体类抗炎药萘普生相当[1]。通过基于竞争活性的蛋白质谱分析(ABPP)研究证实,在用1和10mg/kg po的PF-04457845处理的小鼠中,FAAH被完全抑制[2]。 |
| 激酶实验 | 测定PF-04457845抑制hFAAH和rFAAH的IC 50值。将PF-04457845与FAAH预孵育60分钟,然后通过加入底物油酰胺开始反应。制备小鼠和人体组织,并通过基于竞争活性的蛋白质分析评估抑制剂选择性[1]。 |
| 动物实验 | 将大鼠[1] PF-04457845以指定剂量(mg / kg)口服给予雄性Sprague-Dawley大鼠(200μg-250g),作为2%聚乙烯吡咯烷酮和0.15%十二烷基硫酸钠的H 2 O中的纳米晶体悬浮液。剂量体积为10mL / kg。在给药后4小时评估爪退缩阈值(PWT)。平均PWT测量值,并使用方差分析和非配对T检验进行组间的统计比较。小鼠[2]雄性C57BL6 / J小鼠(7周龄; n = 8)用PF-04457845(1或10mg / kg在聚乙二醇300载体中通过口服给药,体积为4mL / kg)处理,合成大麻素激动剂WIN 55,212-2(1或10mg / kg,18:1:1盐水/ Emulphor /乙醇载体,通过腹膜内给药,体积为10mL / kg),或相应的载体。分别在PF-04457845或WIN 55,212-2施用后4小时或30分钟使用四分体测试评估小鼠的运动不足,体温过低,镇痛作用和僵住作用,除了评估强直性昏厥60秒而不是10秒。使用Student t检验进行统计分析,将每个治疗组与载体进行比较。 |
| 参考文献 |
| 分子式 | C23H20F3N5O2 |
|---|---|
| 分子量 | 455.43200 |
| 精确质量 | 455.15700 |
| PSA | 83.47000 |
| LogP | 4.75380 |
| 储存条件 | 室温,干燥,密封 |
| 危害码 (欧洲) | Xi |
|---|---|
| 危险品运输编码 | NONH for all modes of transport |
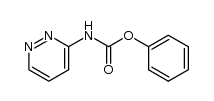
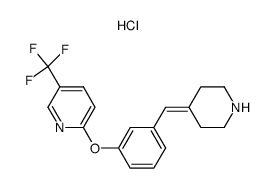
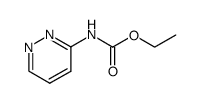
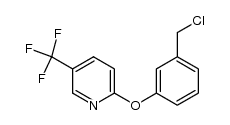

| 上游产品 6 | |
|---|---|
| 下游产品 0 | |