1020315-31-4

1020315-31-4 structure
- Name: PF-04457845
- Chemical Name: N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide
- CAS Number: 1020315-31-4
- Molecular Formula: C23H20F3N5O2
- Molecular Weight: 455.43200
- Catalog: Signaling Pathways Metabolic Enzyme/Protease FAAH
- Create Date: 2016-03-16 08:03:55
- Modify Date: 2024-01-08 13:03:44
-
PF-04457845 is a highly efficacious and selective FAAH inhibitor with IC50 values is 7.2±0.63 nM and 7.4±0.62 nM for hFAAH and rFAAH, respectively.
| Name | N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide |
|---|---|
| Synonyms |
CS-0868
N-pyridazin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-yl]oxy}benzylidene)piperidine-1-carboxamide UNII-H4C81M8YYW PF-04457845 |
| Description | PF-04457845 is a highly efficacious and selective FAAH inhibitor with IC50 values is 7.2±0.63 nM and 7.4±0.62 nM for hFAAH and rFAAH, respectively. |
|---|---|
| Related Catalog | |
| Target |
IC50: 7.2±0.63 nM (hFAAH), 7.4±0.62 nM (rFAAH)[1] |
| In Vitro | PF-04457845 inhibits FAAH by a covalent, irreversible mechanism involving carbamylation of the active-site serine nucleophile of FAAH with high in vitro potency (kinact/Ki and IC50 values of 40300 M-1s-1 and 7.2 nM, respectively, for human FAAH). PF-04457845 has exquisite selectivity for FAAH relative to other members of the serine hydrolase superfamily as demonstrated by competitive activity-based protein profiling. PF-04457845 completely inhibits FAAH in human and mouse membrane proteomes at both 10 and 100 μM with no off targets[1]. PF-04457845 is completely selective for FAAH, and none of the other FP-reactive serine hydrolases in the tested tissues are inhibited by PF-04457845 even at 100 μM[2]. |
| In Vivo | Oral administration of PF-04457845 at 0.1 mg/kg results in efficacy comparable to that of naproxen at 10 mg/kg in a rat model of inflammatory pain. Oral administration of PF-04457845 causes a significant inhibition of mechanical allodynia measured after 4 h with a minimum effective dose (MED) of 0.1 mg/kg. Furthermore, at 0.1 mg/kg (p.o.), PF-04457845 inhibits the pain response to a comparable degree as the nonsteroidal anti-inflammatory drug naproxen at 10 mg/kg[1]. FAAH is confirmed to be completely inhibited in mice treated with PF-04457845 at 1 and 10 mg/kg p.o. by competitive activity-based protein profiling (ABPP) study[2]. |
| Kinase Assay | The IC50 values for the inhibition of hFAAH and rFAAH by PF-04457845 is determined. PF-04457845 is preincubated with FAAH for 60 min before initiating the reaction by the addition of the substrate oleamide. Mouse and human tissues are prepared and inhibitor selectivity is assessed by competitive activity-based protein profiling[1]. |
| Animal Admin | Rats[1] PF-04457845 is administered orally to male Sprague-Dawley rats (200g-250g) at the indicated dose (mg/kg) as a nanocrystalline suspension in 2% polyvinylpyrrolidone and 0.15% sodium dodecyl sulfate in H2O. The dose volume is 10 mL/kg. The Paw Withdrawal Threshold (PWT) is evaluated at 4 h post dose. PWT measurements are averaged and statistical comparisons between groups are made using analysis of variance and unpaired T-tests. Mice[2] Male C57BL6/J mice (7 weeks old; n=8) are treated with PF-04457845 (1 or 10 mg/kg in polyethyleneglycol 300 vehicle by oral administration in a volume of 4 mL/kg), the synthetic cannabinoid agonist WIN 55,212-2 (1 or 10 mg/kg in 18:1:1 saline/Emulphor/ethanol vehicle by intraperitoneal administration in a volume of 10 mL/kg), or the corresponding vehicle. Mice are evaluated for hypomotility, hypothermia, antinociceptive, and cataleptic effects at 4 h or 30 min after PF-04457845 or WIN 55,212-2 administration, respectively, using the tetrad tests except that catalepsy is assessed for 60 s instead of 10 s. Statistical analysis is performed using the Student's t test comparing each treatment group with vehicle. |
| References |
| Molecular Formula | C23H20F3N5O2 |
|---|---|
| Molecular Weight | 455.43200 |
| Exact Mass | 455.15700 |
| PSA | 83.47000 |
| LogP | 4.75380 |
| Hazard Codes | Xi |
|---|---|
| RIDADR | NONH for all modes of transport |
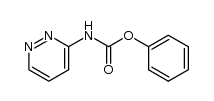
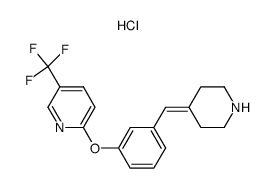
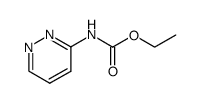
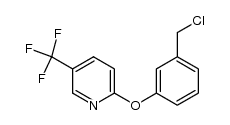

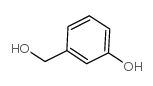
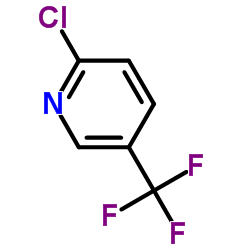
| Precursor 6 | |
|---|---|
| DownStream 0 | |