TAK-733
更新时间:2025-08-20 14:39:01
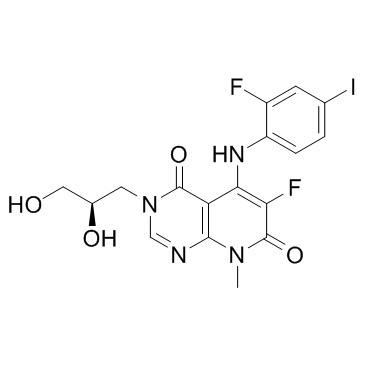
TAK-733结构式

|
常用名 | TAK-733 | 英文名 | TAK-733 |
|---|---|---|---|---|
| CAS号 | 1035555-63-5 | 分子量 | 504.227 | |
| 密度 | 1.9±0.1 g/cm3 | 沸点 | 530.5±60.0 °C at 760 mmHg | |
| 分子式 | C17H15F2IN4O4 | 熔点 | N/A | |
| MSDS | N/A | 闪点 | 274.6±32.9 °C |
TAK-733用途TAK-733 是一种有效的选择性 MEK 抑制剂,IC50 为 3.2 nM。 |

| 中文名 | TAK-733 |
|---|---|
| 英文名 | 3-[(2R)-2,3-dihydroxypropyl]-6-fluoro-5-(2-fluoro-4-iodoanilino)-8-methylpyrido[2,3-d]pyrimidine-4,7-dione |
| 英文别名 | 更多 |
| 描述 | TAK-733 是一种有效的选择性 MEK 抑制剂,IC50 为 3.2 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
MEK:3.2 nM (IC50) |
| 体外研究 | TAK-733表现出有效的酶促和细胞活性,对组成型活性MEK酶的IC50为3.2nM,对细胞中的ERK磷酸化的EC50为1.9nM。 TAK-733不抑制任何其他激酶,受体或离子通道,其抑制剂浓度高达10μM。发现TAK-733适度结合血浆蛋白(人类约97%,小鼠约96%),并且在物种间表现出高渗透性和高微粒体稳定性。它不会抑制高达30μM的P450 [1]。 TAK-733在大多数黑素瘤细胞系中表现出广泛的活性,在体外IC 50>0.1μM时观察到相对抗性。将34个黑素瘤细胞系体外暴露于增加浓度的TAK-733 72小时。在34个细胞系中,27个是BRAFV600E突变体,7个是野生型。进行SRB增殖测定,得到的IC50浓度允许将细胞系分层分为三类:相对抗性,中间和高度敏感。根据相差至少10倍的IC50分配相对抗性和高灵敏度的线[2]。 |
| 体内研究 | 在裸鼠,大鼠,狗和猴中评估TAK-733的药代动力学。在所有物种中都观察到低清除率和高口服生物利用度。 TAK-733在人类癌症的小鼠异种移植模型中表现出广泛的抗肿瘤活性,包括黑素瘤,结肠直肠癌,NSCLC,胰腺癌和乳腺癌的模型[1]。每天口服施用1,3,10和30mg/kg TAK-733 14天(第10至23天)导致A375细胞植入小鼠(5只/组)中的肿瘤生长延迟。 TAK-733(35,70,100和160mg/kg)也以每周3天的间歇给药方案显着抑制肿瘤生长2周(第10,13,15,17,20和22天)。在每天施用30mg/kg TAK-733的小鼠和间歇施用160mg/kg TAK-733的小鼠中观察到三个部分消退(PR),60%响应率。在间歇给予70,100和160mg/kg TAK-733的小鼠中也观察到反应,CR(完全消退)和部分消退(PR)。间歇给药方案的肿瘤消退率更为明显;在160mg/kg(57.29%)观察到肿瘤体积的最大减少,而在30mg/kg每天一次最大减少46.97%。在给药的最后一天,在每天一次给药3,10和30mg/kg或35,70,100和160的小鼠中肿瘤生长显着(%T/C,p <0.05,p <0.05)。间歇性mg/kg [2]。 |
| 细胞实验 | 将处于对数生长期的皮肤黑素瘤细胞系转移到具有盖子的96孔平底板中。将含有2000-3000个活细胞的100μL细胞悬浮液接种到每个孔中并孵育过夜,然后用增加浓度的TAK-733(10,20,30,40,50,60,70,80,90和100)暴露。 nM)72小时。给药后,除去培养基,用冷的10%三氯乙酸将细胞固定30分钟。在4°C。然后用水洗涤细胞并在室温下用0.4%SRB染色30分钟,再用1%乙酸洗涤,然后在室温下用10mM tris染色溶解。在平板读数器上测量565nm处的吸光度。细胞增殖曲线来自原始吸光度(OD)数据。统计分析和数据的图形表示使用GraphPad Prism 5.00版[2]。 |
| 动物实验 | 小鼠[2]使用5至6周龄的雌性无胸腺裸鼠。通过使用胰蛋白酶-EDTA从对数中期培养物中收获细胞产生A375人黑素瘤异种移植肿瘤。将悬浮在Hanks平衡盐溶液(HBSS)中的约5×10 6个细胞皮下注射到6-8周龄小鼠的右侧。当每个实验中所有小鼠的肿瘤大小从100到200 mm3进行抗肿瘤效力研究时,口服TAK-733(1 mg / kg或10 mg / kg),药效学(PD)为300到500 mm3研究[2]。 |
| 参考文献 |
| 密度 | 1.9±0.1 g/cm3 |
|---|---|
| 沸点 | 530.5±60.0 °C at 760 mmHg |
| 分子式 | C17H15F2IN4O4 |
| 分子量 | 504.227 |
| 闪点 | 274.6±32.9 °C |
| 精确质量 | 504.010590 |
| PSA | 109.38000 |
| LogP | 0.28 |
| 外观性状 | 粉末 |
| 蒸汽压 | 0.0±1.5 mmHg at 25°C |
| 折射率 | 1.710 |
| 储存条件 | -20℃ |
![(R)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione结构式](https://image.chemsrc.com/caspic/417/1035555-51-1.png)

![5-(4-amino-2-fluorophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione结构式](https://image.chemsrc.com/caspic/002/1035556-26-3.png)
| cc-621 |
| TAK-733 |
| (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione |
| 3-[(2R)-2,3-Dihydroxypropyl]-6-fluoro-5-[(2-fluoro-4-iodophenyl)amino]-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione |
| (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fIuoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione |
| S2617_Selleck |
| CS-1283 |
| Pyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione, 3-[(2R)-2,3-dihydroxypropyl]-6-fluoro-5-[(2-fluoro-4-iodophenyl)amino]-8-methyl- |
| TAK733 |
本网页内容来自不同专业数据源,如对内容有疑义,欢迎联系service1@chemsrc.com。

- 上海化源生化科技有限公司
- 上海市 上海市 普陀区
- 产品名:TAK-733
- 纯度:98.0%
- 规格:
- 联系人:化源网

- 联系电话:133 1186 9306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:TAK-733
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海脉铂医药科技有限公司
- 上海市 上海市 嘉定区
- 产品名:TAK-733
- 产品网址:http://med-bio.cn/product/578.html
- 纯度:98.0%
- 规格:50mg/10mg/5mg/1g
- 联系人:李先生
- 联系电话:18916172912
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:TAK-733
- 产品网址:http://www.aladdin-e.com/zh_cn/T127214.html
- 纯度:96.0%
- 规格:1mg/5mg/1ml/100mg
- 联系人:阿拉丁
- 联系电话:400-620-6333
查看所有供应商和价格请点击: