
- 上海化源生化科技有限公司
- 上海市 上海市 普陀区
- 产品名:Bitopertin
- 纯度:98.0%
- 规格:
- 联系人:徐经理

- 联系电话:133 1186 9306
- 上海源溪生物科技有限公司
- 上海市 上海市 浦东新区
- 产品名:Bitopertin
- 纯度:98.9%
- 规格:1g
- 联系人:赖经理
- 联系电话:13564518121
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:Bitopertin
- 纯度:98.0%
- 规格:10mg/5mg/50mg/1g
- 联系人:阿拉丁
- 联系电话:400-620-6333
查看所有供应商和价格请点击:
845614-11-1
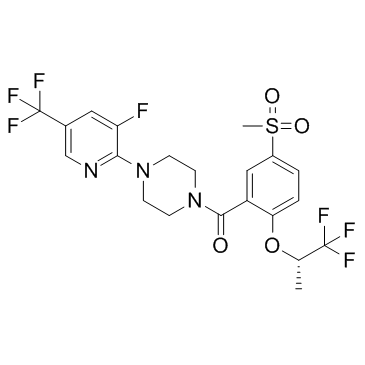
845614-11-1结构式
- 常用中文名:比托派汀
- 常用英文名:Bitopertin
- CAS号:845614-11-1
- 分子式:C21H20F7N3O4S
- 分子量:543.45500
- 相关类别: 信号通路 跨膜转运 GLYT
- 发布时间:2019-01-03 20:08:18
- 更新时间:2025-08-20 16:42:05
-
Bitopertin 是一种有效的非竞争性glycine reuptake 抑制剂,抑制甘氨酸摄取,IC50 为 25 nM。
| 中文名 | (S)-[4-(3-氟-5-三氟甲基吡啶-2-基)哌嗪-1-基][5-(甲磺酰基)-2-(2,2,2-三氟-1-甲基乙氧基)苯基]甲酮 |
|---|---|
| 英文名 | [4-[3-fluoro-5-(trifluoromethyl)pyridin-2-yl]piperazin-1-yl]-[5-methylsulfonyl-2-[(2S)-1,1,1-trifluoropropan-2-yl]oxyphenyl]methanone |
| 中文别名 | 比托派汀 |
| 英文别名 |
[4-(3-fluoro-5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-[5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methyl-ethoxy)-phenyl]-methanone
RG 1678 Paliflutine Paliflutine [INN] (S)-[4-(3-fluoro-5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-[5-methanesulfonyl-2-(2,2,2-trifluoro-1-methyl-ethoxy)-phenyl]-methanone Bitopertin |
| 描述 | Bitopertin 是一种有效的非竞争性glycine reuptake 抑制剂,抑制甘氨酸摄取,IC50 为 25 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: 25 nM (GlyT1)[1] |
| 体外研究 | Bitopertin(RG1678)竞争性地阻断来自中国仓鼠卵巢细胞的膜中人GlyT1b的[3H] ORG24598结合位点。 Bitopertin有效抑制稳定表达hGlyT1b和mGlyT1b的细胞中的[3H]甘氨酸摄取,IC50值分别为25±2nM和22±5nM(n = 6)。相反,Bitopertin对高达30μM浓度的hGlyT2介导的甘氨酸摄取没有影响。 Bitopertin对重组hGlyT1b转运蛋白具有高亲和力。在平衡条件下(室温下1小时),Bitopertin取代[3H] ORG24598结合,Ki为8.1nM。在海马CA1锥体细胞中,Bitopertin在100 nM时增强NMDA依赖性长时程增强,但在300 nM时不增强[1]。另外的分析显示Bitopertin(RG1678)具有优异的针对GlyT2同种型(IC50>30μM)的选择性特征,并且针对包括跨膜和可溶性受体,酶,离子通道和单胺转运蛋白在内的86个靶标组(在41%时抑制<41%)对所有目标测量10μM)[2]。 |
| 体内研究 | Bitopertin(RG1678)剂量依赖性地增加大鼠中通过微透析测量的脑脊髓液和纹状体甘氨酸水平。另外,Bitopertin减弱由精神兴奋剂D-苯异丙胺或NMDA受体甘氨酸位点拮抗剂L-687,414在小鼠中诱导的过度运动。 Bitopertin还可以防止长期用苯环利定(一种NMDA受体开放通道阻滞剂)治疗的大鼠对D-苯异丙胺攻击的过度反应。施用载体对纹状体甘氨酸的细胞外水平没有影响,其在整个实验中保持恒定。相反,口服Bitopertin(1-30mg/kg)产生细胞外甘氨酸水平的剂量依赖性增加。 Bitopertin 30 mg/kg产生的甘氨酸水平比治疗前水平高2.5倍。在药物施用后3小时,与载体处理的动物相比,在用Bitopertin(1-10mg/kg)处理的大鼠的CSF中观察到类似的甘氨酸浓度的剂量依赖性增加。有趣的是,Bitopertin给药后3小时CSF甘氨酸水平增加与微渗透实验在同一时间点的增加非常相似[1]。大鼠和猴子的体内药代动力学研究表明,Bitopertin(RG1678)在两个物种中都具有低血浆清除率,中等分布容量,良好的口服生物利用度(大鼠为78%,猴子为56%),且有利终末半衰期(大鼠5.8小时,猴子6.4小时)。血浆蛋白结合在两种临床前物种(97%)和人体(98%)中很高。 Bitopertin在大鼠(脑/血浆= 0.7)中的CNS渗透性优于小鼠(脑/血浆= 0.5)[2]。 |
| 激酶实验 | 进行[3H] ORG24598与hGlyT1和大鼠脑膜的结合和解离动力学分析。 [3H] ORG24598结合实验使用来自表达hGlyT1b的CHO细胞的膜以及来自小鼠,大鼠,猴和狗脑胶的膜进行。通过将[3H] ORG24598添加至大鼠,小鼠,猴和狗前脑膜(40μg/孔)和细胞膜(10μg/孔),总体积为500μL,在室温下3小时来确定饱和等温线。饱和结合实验通过基于Excel的曲线拟合程序进行分析,该程序使用源自双分子反应方程和质量作用定律的Michaelis-Menten方程:B =(Bmax×[F])/(Kd + [F] ),其中B是平衡时结合的配体量,Bmax是结合位点的最大数量,[F]是游离配体的浓度,Kd是配体解离常数。对于抑制实验,将膜与3nM [3H] ORG24598和10种浓度的Bitopertin在室温下孵育1小时。在浓度增加的[3H] ORG24598(1-300nM)存在下进行Schild分析。如上所述导出IC 50值。 Ki值根据下式计算:Ki = IC50 /(1+ [L] / Kd)[1]。 |
| 动物实验 | 小鼠[1]用Bitopertin(0.3,3,1和10mg / kg口服)或载体(po)处理雄性NMRI小鼠(20-30g)。 1分钟后,给予L-687,414(50mg / kg sc)或载体。在活动室中适应15分钟后,记录水平活动60分钟。还研究了Bitopertin对L-678,414诱导的机能亢进的影响的时间过程;在施用Bitopertin后2.5,4.5和24小时评估运动活性(L-678,414总是在活动程序前15分钟给予)。此外,研究了亚慢性Bitopertin的作用。小鼠连续4天接受载体或Bitopertin(1mg / kg口服)并且在第5天评估L-678,414诱导的活动过度。大鼠[1] Wistar大鼠接受14天的PCP HCl治疗(5mg / kg)或载体(NaCl 0.9%,5mL / kg ip)。最后一次注射后24小时,使大鼠(每组6-18只)单独适应试验盒30分钟。然后大鼠接受Bitopertin(1,3,10mg / kg口服)或载体(聚山梨醇酯80,HEC,对羟基苯甲酸甲酯和对羟基苯甲酸甲酯6.0; 5mL / kg口服),1小时后接受1mg / kg D-苯丙胺或载体ip在给予Bitopertin后直接记录水平活性,直至给予苯丙胺后120分钟。通过ANOVA分析数据,补充Fischer的最小差异事后检验。 |
| 参考文献 |
| 分子式 | C21H20F7N3O4S |
|---|---|
| 分子量 | 543.45500 |
| 精确质量 | 543.10600 |
| PSA | 88.19000 |
| LogP | 5.01870 |
| 储存条件 | -20°C |
|
~79%
845614-11-1 |
| 文献:Pinard, Emmanuel; Alanine, Alexander; Alberati, Daniela; Bender, Markus; Borroni, Edilio; Bourdeaux, Patrick; Brom, Virginie; Burner, Serge; Fischer, Holger; Hainzl, Dominik; Halm, Remy; Hauser, Nicole; Jolidon, Synese; Lengyel, Judith; Marty, Hans-Peter; Meyer, Thierry; Moreau, Jean-Luc; Mory, Roland; Narquizian, Robert; Nettekoven, Mathias; Norcross, Roger D.; Puellmann, Bernd; Schmid, Philipp; Schmitt, Sebastien; Stalder, Henri; Wermuth, Roger; Wettstein, Joseph G.; Zimmerli, Daniel Journal of Medicinal Chemistry, 2010 , vol. 53, # 12 p. 4603 - 4614 |
| 上游产品 2 | |
|---|---|
| 下游产品 0 | |