
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
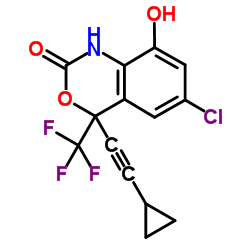
- 产品名:依法韦仑
- 纯度:98.0%
- 规格:1ml/10mg/250mg/1g
- 联系人:阿拉丁

- 联系电话:400-620-6333
- 武汉敬康恩生物医药科技有限公司
- 湖北省 武汉市 东西湖区
- 产品名:Efavirenz
- 纯度:98.0%
- 规格:1g
- 联系人:周经理
- 联系电话:15871494362
查看所有供应商和价格请点击:
154598-52-4

| 中文名 | 依法韦仑 |
|---|---|
| 英文名 | efavirenz |
| 中文别名 |
(4S)-6-氯-4-(环丙乙炔)-4-(三氟甲基)-苯并-1,4-二氢唑-2-酮
依发韦仑 依法韦伦 依非韦伦 |
| 英文别名 |
Stocrin
EFAVIRNEZ Efavirenz (200 mg) (S)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one (4S)-6-Chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-4H-3,1-benzoxazin-2-ol (4S)-6-Chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-1,4-dihydro-2H-3,1-benzoxazin-2-one 4H-3,1-Benzoxazin-2-ol, 6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-, (4S)- DMP 266 SUSTIVA MDP-266 Efavirenz 2H-3,1-Benzoxazin-2-one, 6-chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-, (4S)- |
| 描述 | Efavirenz 是一种有效的野生型 HIV-1 RT 抑制剂,Ki 为 2.93 nM,抑制 HIV-1 复制,IC95 为 1.5 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
Ki: 2.93 nM (HIV-1 RT)[1] |
| 体外研究 | Efavirenz(L-743726)被发现能够抑制95%抑制浓度≤1.5μM的一组非核苷类逆转录酶(RT)抑制剂(NNRTIs)抗性突变病毒,每种病毒均表达一个RT氨基酸酸取代。还测试了依法韦仑对多种聚合酶的活性,发现其无活性(IC50>300μM)。依法韦仑有效抑制几种野生型T淋巴细胞系适应变异体。在初级淋巴细胞和单核细胞样细胞培养物中观察到病毒的野生型初级分离株具有相同的活性(IC95,1.5至3.0nM)。依法韦仑还有效抑制表达RT氨基酸取代的HIV-1变体,其赋予对其他NNRTI的易感性丧失。为了比较[1]。 Efavirenz是一种非核苷类似物逆转录酶抑制剂(NNRTI),IC50为60 nM [2]。 Efavirenz使用RNA PPT引发的底物抑制合成,IC50为17 nM [3]。 |
| 体内研究 | 静脉内给药后,Efavirenz(L-743726)从大鼠中快速清除,但从猴子中清除得相当慢。两个物种的大量分布(体液水量的2至4倍)表明广泛的组织结合。大鼠的口服生物利用度为16%。在猴子中,施用1mg/kg静脉内剂量后依法韦仑的半衰期超过2.5小时。依法韦仑口服吸收良好。给予口服剂量的猴子作为0.5%甲基纤维素水溶液中的细悬浮液,在血浆中产生一致的高水平。 2.0mg/kg剂量在约3.0小时产生0.5μM的峰值水平。绝对生物利用度估计为42%。 10mg/kg剂量在血浆中产生3.22μM的峰值水平。给予单个黑猩猩的10mg/kg口服剂量分别在给药后2,8和24小时给予血浆中4.12,2.95和2.69μM的浓度[1]。 |
| 激酶实验 | 表达,纯化重组RT酶,并评估Efavirenz(L-743726)的抑制作用。测定每种测试酶的Ki和Kii值。野生型RT仅显示非竞争性抑制动力学(数据未显示),因此Ki和Kii值相同。对于突变酶,不假设纯的非竞争性抑制,因此Ki和Kii的值都是从线性混合型抑制方程获得的。 Ki和Kii值之间的两到三倍差异可能反映了突变体RTs的竞争性抑制的一小部分[1]。 |
| 动物实验 | 小鼠[1]研究在大鼠,恒河猴和单只黑猩猩中进行。对于静脉内给予大鼠的药物的分析(iv),一组(n = 4或5)禁食的雄性Sprague-Dawley大鼠(体重250至450g)接受推注(体积为1mL / kg体重)通过植入右颈静脉的套管将Efavirenz在DMSO中的重量)。对于口服研究,通过使用在0.5%甲基纤维素水溶液中制备的依法韦仑的悬浮液通过管饲法给予大鼠。类似地,四只猴子通过隐静脉以0.1mL / kg的体积接受化合物在DMSO中的静脉推注,或者通过使用鼻胃管以悬浮液的形式口服施用化合物。在给药前,猴子禁食18小时。通过使用化合物的水悬浮液通过自愿摄取口服给药一只非麻醉的,非禁食的雄性黑猩猩(体重,约60kg)。在所有研究中,在适当的时间获得肝素化血液。通过离心立即分离血浆并储存在-20℃直至分析。血浆样品用二氯甲烷萃取;然后通过高效液相色谱分析。 |
| 参考文献 |
| 密度 | 1.5±0.1 g/cm3 |
|---|---|
| 沸点 | 422.7±55.0 °C at 760 mmHg |
| 熔点 | 139-141ºC |
| 分子式 | C14H9ClF3NO2 |
| 分子量 | 315.675 |
| 闪点 | 209.4±31.5 °C |
| 精确质量 | 315.027405 |
| PSA | 38.33000 |
| LogP | 3.72 |
| 外观性状 | 白色至略粉红色结晶粉末 |
| 蒸汽压 | 0.0±1.1 mmHg at 25°C |
| 折射率 | 1.582 |
| 储存条件 | <0°C;避免加热 |
| 水溶解性 | 水溶性:不溶;易溶于:甲醇,乙醇 |
| 分子结构 | 1、 摩尔折射率:68.40 2、 摩尔体积(cm3/mol):205.2 3、 等张比容(90.2K):549.5 4、 表面张力(dyne/cm):51.3 5、 极化率(10-24cm3):27.11 |
| 计算化学 | 1.疏水参数计算参考值(XlogP):4 2.氢键供体数量:1 3.氢键受体数量:5 4.可旋转化学键数量:1 5.互变异构体数量:2 6.拓扑分子极性表面积38.3 7.重原子数量:21 8.表面电荷:0 9.复杂度:519 10.同位素原子数量:0 11.确定原子立构中心数量:1 12.不确定原子立构中心数量:0 13.确定化学键立构中心数量:0 14.不确定化学键立构中心数量:0 15.共价键单元数量:1 |
| 更多 | 1.性状:白色或类白色结晶粉末 |
| 符号 |

GHS09 |
|---|---|
| 信号词 | Warning |
| 危害声明 | H410 |
| 警示性声明 | P273 |
| 危害码 (欧洲) | N |
| 风险声明 (欧洲) | 50 |
| 安全声明 (欧洲) | 61 |
| 危险品运输编码 | UN3082 - class 9 - PG 3 - DOT NA1993 - Environmentally hazardous substances, liquid, n.o.s. HI: all (not BR) |
| RTECS号 | DM3440000 |
| 海关编码 | 2942000000 |
| 上游产品 9 | |
|---|---|
| 下游产品 2 | |
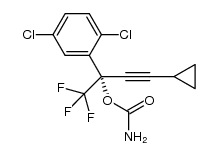
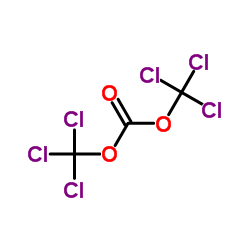
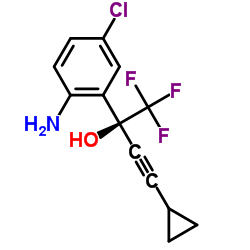
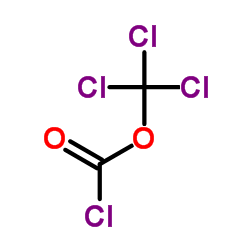

![(S)-2-(5-chloro-2-((1S,4R)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxamido)phenyl)-4-cyclopropyl-1,1,1-trifluorobut-3-yn-2-yl 1-chloroethyl carbonate结构式](https://image.chemsrc.com/caspic/263/1262030-22-7.png)
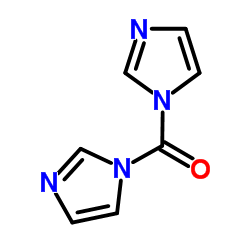
1.对氯苯胺溶于氯仿和饱和碳酸钠水溶液,滴加2,2-二甲基丙酰氯。加毕,在室温搅拌,过滤收集固体,滤液分层,分出的氯仿层用盐水洗,干燥,浓缩。浓缩得到的物质和上述得到的固体一起用煮沸的乙酸乙酯己烷重结晶,得N-2,2-二甲基丙酰基对氯苯胺。将该酰化物溶于四氢呋喃,滴加正丁基锂的己烷溶液,搅拌,再滴加三氟乙酸乙酯,加入盐酸和乙酸乙酯,分出有机层,用盐水洗涤,干燥,减压浓缩,得到的物质在盐酸中回流,冷却,加入乙酸乙酯,用浓氨水调至碱性。分出有机层,用盐水洗涤,干燥,浓缩,得到2-三氟乙酰基-4-氯苯胺。将环丙基乙炔溶于四氢呋喃,滴加乙基溴化镁的乙醚溶液进行反应,完毕后,加入2-三氟乙酰基-4-氯苯胺,再滴加饱和的氯化铵水溶液。用乙酸乙酯萃取,萃取液合并后用盐水洗,干燥,浓缩,得烷化产物。将其和羰基二咪唑溶于无水四氢呋喃。然后减压蒸出溶剂,加入乙酸乙酯和水。水层用乙酸乙酯萃取。萃取液和有机层合并,用盐酸、饱和碳酸氢钠和盐水洗涤,经干燥、减压浓缩,得到消旋的依非韦仑。将其和(-)樟脑酰氯在二氯甲烷反应,然后在正丁醇中,加入盐酸反应,拆分得光学活性的依非韦仑。
2.
合成文献较多,见文献[6-12].推荐文献[6,7]的路线和方法
(1)N-(4-氯苯基)-2,2-二甲基丙酰胺(041-2)的制备
在反应瓶中加入对氯苯胺(041-4)127.57g(1mol)、氯仿1200ml和饱和的Na2CO3水溶液1200ml,搅拌混合,于1h内滴加2,2-二甲基丙酰氯129ml(1.05mol),滴加过程中应剧烈搅拌,滴毕,反应混合物于环境温度下继续搅拌反应23h.析出白色晶体过滤收集,滤液转至分液漏,分取有机层(氯仿层),依次用水和盐水洗涤,无水MgSO4干燥,过滤,滤液减压蒸除溶剂,剩余物与上述过滤出的固体合并,用煮沸的乙酸乙酯/己烷重结晶,得化合物041-2 185.6g,收率87.8%,
该化合物为白色结晶性固体.
(2).1-(2-氨基-5-氯苯基)-2,2,2-三氟乙酮(041-3)的制备
往预先干燥好的反应瓶中加入上步制备的化合物041-2 100g(472mmol)和干燥的THF 1000ml,于搅拌下用冰浴冷却至0 ºC,于该温度下滴加正丁基锂的己烷溶液(2.5mol/L)387ml(968mmol),滴加过程中保持内温低于5 ºC,1h内滴加毕,在0 ºC拌反应1h.在保持反应过程中,反应液中产生橙色沉淀.向该混合物中再滴加1,1,1-三氟乙酸乙酯115ml(968mmol),于1h加完,加毕,继续搅拌30min.加入5%盐酸适量终止反应.反应混合物中加入乙酸乙酯1000ml释,搅拌充分后静置分层.分取有机层用盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,得160g黄色油状物.将其悬浮于3mol/L盐酸1000ml中.溶液搅拌回流24h.冷却,加入乙酸乙酯1000ml稀释,混合物用浓氨水调至PH呈碱性.静置分层,取有机层用盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,浓缩液经硅胶柱(1.5kg硅胶)色谱分离(洗脱剂:15%乙酸乙酯的己烷溶液),含目的物的洗脱液减压浓缩至干,得到的固体用煮沸的己烷重结晶,得化合物041-3 57g,率54%,为亮黄色晶体,mp91~92 ºC(纯品)。
(3)。2-(2-氨基-5-氯苯基)-4-环丙基-1,1,1-三氟-3-丁炔-2-醇(041-5)的制备
在反应瓶中加入THF 250ml和环丙基乙炔(041-4)23g(0.348mol),搅拌溶解,于1h内搅拌下
滴加3mol/L乙基溴化镁的乙醚溶液116ml(0.348mol).加毕,于0 ºC 拌反应1h.然后在40 ºC 搅拌反应3h.将反应液再冷却至0 ºC ,在5min内往该反应液中分批加入上步制备的化合物041-3 15.56g(0.0696mol),加毕,在0 ºC 拌反应1.5h.再在0 ºC 滴加饱和的氯化铵水溶液700ml终止反应.反应混合物用乙酸乙酯(400ml*2)提取,合并有机相,用盐水洗涤,无水MgSO4干燥,过
滤,滤液经浓缩得到黄色固体,用煮沸的己烷重结晶(沸己烷最终体积100ml),得14.67g,母液浓缩处理可回收041-5 2.1g,合计得041-5 16.77g,mp153~154 ºC .
(4).(+-)-6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2H-3,1-苯并嗪-2-酮(041-6)的制备
在反应瓶中加入上步制备的化合物041-5 15.00g(0.0518mol)和1,1’-羰基二咪唑(CDD)41.98g(0.259mol)的干燥的THF250ml溶液,在氩气保护下于55 ºC 搅拌反应0C;$利用旋转蒸发器将反应液中溶剂减压蒸除,剩余物加入乙酸乙酯500ml和水400ml,分搅拌后静置分层.水层用乙酸乙酯再提取一次,合并提取液和有机层,用2%盐酸(200ml*2)洗涤,饱和碳酸氢钠水溶液洗涤,最后用盐水洗涤.无水硫酸镁干燥,过滤,滤液减压浓缩回收溶剂,得到剩余物16.42g,固体,将其用乙酸乙酯/己烷重结晶得041-6 12.97g品,为白色结晶,mp178~180 ºC .
(5)6-氯-1-(1S)-樟脑酰基-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2H-3,1-苯并嗪-2-酮(041-7)的
制备
在反应瓶中加入上步制备的化合物041-6 (即消旋的Efavirenz)12.97g(0.041mol)、4-二甲基氨基吡啶(DMAP)1.02g(0.0083mol)和(-)樟脑酰氯[(-)-camphanic acid chloride]14.22g(0.06556mol)的干燥二氯甲烷350ml溶液,搅拌,在冰浴冷却下和氩气保护下加入三乙胺22.84ml(0.164mol).移去冰浴,自然升至室温,搅拌反应75min(TLC跟踪,用4%的乙酸乙酯的氯仿溶液作展开剂,点样展开,原料斑点消失,反应则基本完成).将反应液用氯仿500ml稀释,然后用10%的柠檬酸洗涤2次,用水洗1次,盐水洗1次.无水MgSO4干燥,过滤,滤液减压浓缩回收溶剂,剩余物为无色泡沫状物,加入己烷200ml煮沸并研碎,冷却至室温,过滤,滤饼用少量冷己烷洗涤,抽干,真空干燥,得041-7 7.79g,白色晶体,mp164~165 ºC纯度99.2%(HPLC法)
(6)。(4S)-6-氯-4-(环丙基乙炔基)-1,4-二氢-4-(三氟甲基)-2H-3,1-苯并嗪-2-酮(艾法韦瑞)(041)的合成
在反应瓶中加入上步制备的化合物041-7 7.5g(0.01512mol)和正丁醇150ml,在氩气保护下升温至60ºC搅拌溶解,再加入1mol/L盐酸10ml,仍保持反应混合物在60ºC,搅拌反应72h.用NaHCO3水溶液中和,真空蒸除正丁醇.剩余物用THF150ml溶解,加入2mol/L氢氧化锂(LiOH)50ml,于室温搅拌3h.用乙酸乙酯稀释,搅拌充分后用水洗2次,用盐水洗涤1次,无水MgSO4干燥,过滤,滤液减压蒸除溶剂,得白色固体,将其用热己烷重结晶,得041 3.43g,为白色晶体,mp131~132ºC

| 海关编码 | 2942000000 |
|---|